
氨酚咖匹林片中对氨基酚和对氯苯乙酰胺的检测
2021-05-21 06:21吴毅彦郝桂明
天津药学 2021年2期
程 侠,吴毅彦,郝桂明,王 卫
(天津市药品检验研究院,天津 300070)
氨酚咖匹林片的主要成分为对乙酰氨基酚、咖啡因和阿司匹林,是一种解热镇痛药的非处方药,对氯苯乙酰胺和对氨基酚是对乙酰氨基酚原料药含有的两个主要杂质,均是对乙酰氨基酚合成过程中引入的杂质[1],毒性较大[2-3]。为了保证药品的安全,有必要控制对氯苯乙酰胺和对氨基酚的含量,而氨酚咖匹林片现行质量标准[国家食品药品监督管理总局国家药品标准WS1-10001(HD-1126)-2002-2009]和相关文献中均没有检测这两个杂质含量的方法。本试验建立了一个HPLC 方法同时测定对氯苯乙酰胺和对氨基酚的含量,旨在提高氨酚咖匹林片的质量控制技术。
1 仪器与试药
1.1 仪器 Waters2695 高效液相色谱仪,配有自动进样器、2998PDA 检测器、Empower 色谱工作站(美国Waters 公司);Mettler XPE105 电子天平(瑞士梅特勒公司)。
1.2 试药 对氯苯乙酰胺对照品(中国食品药品检定研究院,批号 100850-201803,含量:100%),对氨基酚对照品(中国食品药品检定研究院,批号:100802-201203,含量:100%);7 批氨酚咖匹林片样品为抽验样品,规格:阿司匹林0.226 8 g,咖啡因33.4 mg,对乙酰氨基酚 0.136 g(厂家 A,批号 2010080、2010082、2011086、2010081、2007077、2006069、2001009);甲 醇(色谱纯,默克公司);水为超纯水;磷酸氢二钠、磷酸二氢钠、25%四丁基氢氧化铵溶液均为市售分析纯。
2 方法与结果
2.1 色谱条件 色谱柱:ACCHROM XUnion C8(250 mm×4.6 mm,5 μm);流动相 A 为磷酸盐缓冲液(取磷酸氢二钠8.95 g,磷酸二氢钠3.9 g,加水溶解至1 000 ml,加25%四丁基氢氧化铵溶液4.8 ml),流动相B 为甲醇,梯度洗脱程序见表1;柱温:40 ℃;流速:1.0 ml/min;进样量:20 μl;检测波长:245 nm。

表1 梯度洗脱程序
2.2 溶液制备
2.2.1 混合对照品贮备液 精密称取对氯苯乙酰胺对照品10.06 mg 和对氨基酚对照品20.19 mg 分别置20 0 ml 和 20 ml 量瓶中,分别加溶剂[水-甲醇(60∶40)]适量,振摇10 min,用溶剂稀释至刻度,摇匀,分别作为对氯苯乙酰胺和对氨基酚对照品贮备液;精密量取上述2 种对照品贮备液各10 ml 置同一100 ml 量瓶中,加溶剂稀释至刻度,摇匀,作为混合对照品贮备液。
2.2.2 混合对照品溶液 精密量取混合对照品贮备液10 ml 置100 ml 量瓶中,加溶剂稀释至刻度,摇匀,作为混合对照品溶液(约含对氯苯乙酰胺0.5 μg/ml,对氨基酚 10 μg/ml)。
2.2.3 供试品溶液 精密称取本品细粉适量(约相当于对乙酰氨基酚0.2 g),置20 ml 量瓶中,加溶剂10 ml,振摇10 min,加溶剂稀释至刻度,摇匀,滤过,取续滤液,即得供试品溶液。
2.2.4 供试品加标溶液 精密称取本品细粉适量(约相当于对乙酰氨基酚0.2 g),置20 ml 量瓶中,精密加入混合对照品贮备液2.0 ml,加溶剂10 ml,振摇10 min,加溶剂稀释至刻度,摇匀,滤过,取续滤液,即得供试品加标溶液。
2.2.5 空白溶液 水-甲醇(60∶40)溶液。
2.3 专属性试验 取空白溶液、混合对照品溶液、供试品加标溶液及供试品溶液,按上述色谱条件进行测定,记录色谱图。对照品色谱图中对氨基酚峰和对氯苯乙酰胺峰的理论板数分别为10 287 和118 364;供试品加标溶液色谱图中对氨基酚峰和对氯苯乙酰胺峰与各自相邻峰均能分离良好,其中对氨基酚峰与其相邻杂质峰之间的分离度为2.2,对氯苯乙酰胺峰与其相邻杂质峰的分离度为3.3;空白无干扰。色谱图见图1。

图1 空白对照(A)混合对照品(B)供试品加标(C)供试品(D)HPLC 色谱图
2.4 线性关系与范围 分别精密量取混合对照品贮备液 0.1、0.5、1.0、5.0、10.0 和 12.5 分别置 50 ml 量瓶中,加溶剂稀释至刻度,摇匀,作为对照品系列溶液,按上述色谱条件测定,记录色谱图,以浓度C 为横坐标,峰面积A 为纵坐标,绘制标准曲线,得到线性方程和相关系数如下:对氯苯乙酰胺A=110 396 C-1 568.1(r=0.999 9);对氨基酚 A=35 763 C-3 040.5(r=0.999 9)。结果表明:对氯苯乙酰胺和对氨基酚分别在0.05~1.26 μg/ml 和 0.20~25.24 μg/ml 的范围内线性关系良好。
2.5 检测限 取混合对照品贮备溶液,分别用溶剂逐级稀释,按上述色谱条件进行测定,记录色谱图,以信噪比S/N 值达到3 左右时计算检测限,结果对氯苯乙酰胺为 0.025 μg/ml,对氨基酚为 0.01 μg/ml。
2.6 精密度试验 取混合对照品溶液连续进样6 次,计算对氯苯乙酰胺和对氨基酚峰面积的RSD 分别为0.2%和0.4%(n=6),表明该方法精密度良好。
2.7 重复性试验 取氨酚咖匹林片样品(批号2007077)细粉适量(约相当于对乙酰氨基酚0.2 g)6 份,置20 ml量瓶中,按“2.2.3”项下方法配制供试品溶液,分别进样,按外标法以峰面积计算含量,结果对氨基酚的含量为0.001 9%,RSD 为0.84%,对氯苯乙酰胺未检出,表明该方法重复性良好。
2.8 稳定性试验 取混合对照品溶液,放置0、1、2、4、6、8、10 和 12 h 后,按上述色谱条件测定,对氯苯乙酰胺和对氨基酚峰面积的RSD 分别为0.46%和1.78%(n=6),对氯苯乙酰胺在12 h 内比较稳定,但对氨基酚在4 h 后开始出现降解现象。因此,溶液配制好后应于低温保存并尽快进样分析[4-5]。
2.9 回收率试验 精密称取已知含量的氨酚咖匹林片样品(批号2010080)细粉适量(约相当于对乙酰氨基酚0.2 g)9 份,置20 ml 量瓶中,分别加入混合对照品贮备液 1.6、2.0 和 2.4 ml,每个浓度各 3 份,按“2.2.3”项下方法分别配制溶液,分别进样,结果对氨基酚平均回收率为100.34%,RSD 为0.63%;对氯苯乙酰胺平均回收率为100.72%,RSD 为1.35%。见表2 和表3。

表2 对氨基酚回收率试验结果(n=9)
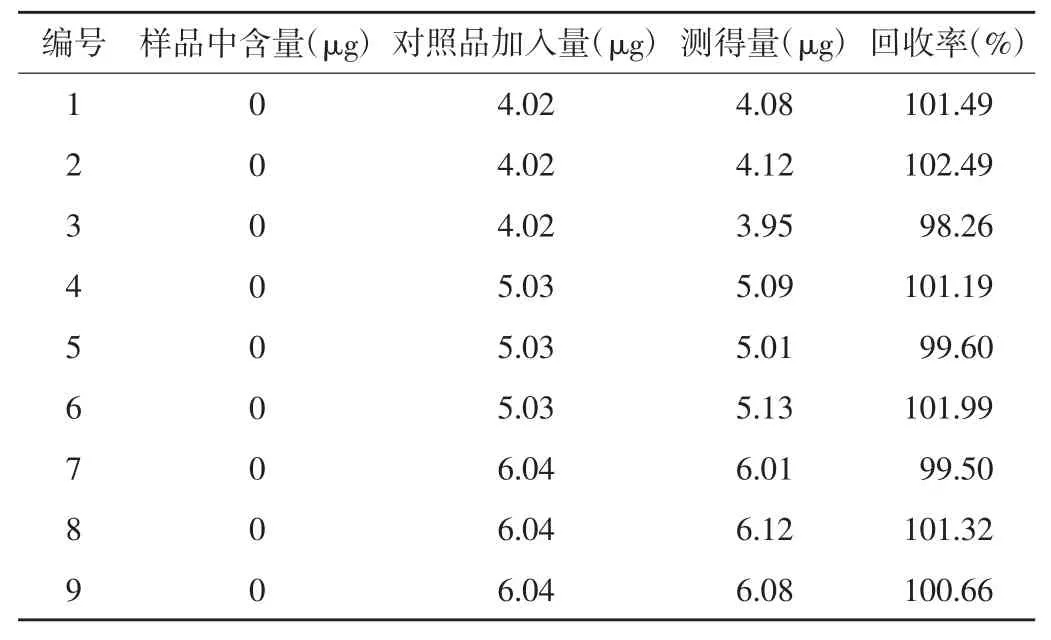
表3 对氯苯乙酰胺回收率试验结果(n=9)
2.10 样品测定 对7 批样品,按“2.2.3”项下方法分别配制供试品溶液,另取混合对照品溶液,分别进样测定,并按外标法以峰面积分别计算两个杂质的含量,结果见表4。对氨基酚的含量测定结果均在其浓度线性范围内。

表4 样品测定结果(n=2)
3 讨论
3.1 色谱条件的选择 《中国药典》2015 年版二部中收载的对乙酰氨基酚原料药[6]中“对氯苯乙酰胺”和“对氨基酚及有关物质”的测定方法采用了两个色谱系统来测定这两个杂质的含量,工作效率低且成本高。本方法在《中国药典》标准的基础上优化了色谱条件,采用梯度洗脱的方法,在较短时间内同时测定对氯苯乙酰胺和对氨基酚的含量,且在测定的7 批样品色谱图中对氯苯乙酰胺和对氨基酚均能与相邻的杂质峰得到很好的分离。此方法专属性好,线性、精密度和回收率良好,可快速测定氨酚咖匹林片中的对氯苯乙酰胺和对氨基酚的含量。
3.2 溶液稳定性 此次考查结果表明,常温下对照品溶液中对氨基酚峰在4 h 后开始出现降解趋势,虽然对氨基酚峰的峰面积在12 h 内的RSD<2.0%,仍建议溶液配制好后于低温保存并尽快进样分析。
3.3 样品的测定 《中国药典》2015 年版二部中收载的对乙酰氨基酚原料药[6]中对氯苯乙酰胺和对氨基酚的限度均为0.005%,收载的对乙酰氨基酚片[6]中对氯苯乙酰胺的限度为0.005%,对氨基酚的限度为0.1%。此次考查的7 批样品中,对氯苯乙酰胺均未检出,其中有3 批检出对氨基酚,但含量均小于0.05%,说明这7批样品对这两个杂质的质量控制得较好。
猜你喜欢
化工进展(2022年9期)2022-10-13
农药学学报(2022年3期)2022-06-14
保健与生活(2022年12期)2022-06-09
节能与环保(2022年3期)2022-04-26
安徽农学通报(2022年6期)2022-04-07
保健与生活(2020年4期)2020-03-02
家庭科学·新健康(2018年11期)2018-11-16
上海预防医学(2017年1期)2017-03-09
发明与创新·中学生(2017年1期)2017-01-20
家庭用药(2016年8期)2016-05-14
