
G蛋白偶联雌激素受体研究进展及在食品功能评价中的应用
2021-05-19 02:22鲁丁强庞广昌
食品科学 2021年7期
鲁丁强,庞广昌
(天津商业大学生物技术与食品科学学院,天津市食品生物技术重点实验室,天津 300134)
大量研究和综述发现雌激素不仅在女性,也在男性发育、分化、成熟、生殖、健康与疾病中发挥重要作用[1]。外源性雌激素,即所谓的植物雌激素涵盖了几乎所有的食物和中草药有效成分,在健康和疾病中的作用被越来越多的研究所揭示[2]。最早在1926年,发现植物化学物在动物中发挥和内源性雌激素平行的生物学作用[3-4]。1931年报道了富含异黄酮的大豆类食品所具有的健康作用正是由于含有植物雌激素类化合物——大豆异黄酮[5]。1938年,Zondek等[6]报道香精油中的一些化合物具有和内源性雌激素相似的结构和功能,并得到了Bennets等[7]的证实。Axelson等[8]发现澳大利亚绵羊长期啃食三叶草造成不育,主要是因为这种草中含有雌马酚。1980年代,在尿液中也分离鉴定到了植物雌激素[8],并通过一系列的流行病学和生理生化研究证明,异黄酮与内源性雌激素受体(estrogen receptor,ER)α和ERβ具有亲和作用[9]。此后发现,大多数植物雌激素都可能通过ERα和ERβ这两种经典的内源性ERs发挥作用[10]。Barton等[11]在回顾G蛋白偶联雌激素受体(G-protein coupled estrogen receptor,GPER)发现20 周年时提出:ERα和ERβ作为固醇类受体,其起源和进化可以追溯到5亿 年前,而G蛋白偶联受体(G-protein coupled receptors,GPCRs)则可以追溯到10亿 年前,明显早于前者。而且,早在1939年,Reynolds等[12]就发现雌激素对女性的作用存在一条通过神经系统传导的快速途径,而这两种经典的ERs本质上属于配体介导的转录因子激活(或抑制),从而改变众多基因表达,属于基因组学途径。显然,从逻辑上来推论,这两种ERs之上一定还存在一条更古老的非基因组学的、快速的膜结合受体。最早发现一种可能具有传递雌激素信号的GPCR被命名为GPR30,属于孤儿受体(尚未确定其功能的GPCR归类为孤儿受体);到2000年,积累的证据表明,GPR30也是雌激素快速“非基因组学”信号传递所必需的,因此GPR30获得了雌激素研究领域的广泛关注[12]。2005—2006年,大量实验发现,雌激素与在细胞膜上表达的GPR30结合传递非基因组信号,与此同时,激活ERα和ERβ核转录活性,并鉴定分离了多种GPR30选择性激活剂或拮抗剂。到2007年,有关GPR30的论文已经达到1 000 篇以上,促使国际药理学联合会重新命名GPR30为GPER。这是因为GPER的发现和研究为药物筛选、内源性和外源性雌激素的功能评价、信号途径及其与糖尿病、肥胖、代谢综合征、癌症的发生、发展、病情预测、预防和治疗等领域打开了一扇大门。
有关GPER的综述已有很多,但集中在生物和医药领域,而有关食品功能评价方面的研究还鲜见报道。目前,所研究的功能性食品以及食品安全检测与控制领域,绝大多数都与ER、芳香烃受体(aryl hydrocarbon receptor,AHR)有关[13],这些受体主要是通过基因组途径调节基因表达,需要经过数小时,甚至数天的时间,但需要强调的是,食品功能性成分或毒素发挥作用显然具有快速信号途径,如食欲、营养吸收、神经、代谢、免疫、内分泌控制需要GPER的快速控制途径。可见GPER所控制的非基因组学快速途径才是控制雌激素类化合物发挥生理功能的主体途径,同时也是控制基因组学途径的枢纽,本文将就这些问题进行综合分析。
1 胃肠道是植物化学物发挥作用的主要部位
越来越多的研究结果显示,无论是中草药还是植物化学物,都不大可能直接进入体内发挥作用,即使极少量化合物进入血液循环系统也必将很快被代谢分解或经肝脏解毒酶分解后经尿液排出,不可能发挥主体生理作用。可见,一个不可回避的问题是,作为中草药和植物化学物到底是如何发挥生理、病理、甚至毒性作用的?Pang Guangchang等[14]认为,食品和中草药主要是通过服用到胃、肠道中发挥作用,而不是通过吸收到循环系统中发挥作用;其具体机制为:胃肠系统,特别是肠黏膜系统极其丰富多样的受体构成机体对食入的所有外来化合物分子结构的识别系统,随时随地向机体传递神经、内分泌、代谢、免疫等电化学信号。这个系统不仅可以传感机体所必需的营养(即宏营养)信号,而且可以识别微营养、抗营养、毒素、植物化学物、中草药等化学成分,从而控制营养吸收、合成与分解代谢、能量储存与利用、免疫防御以及生理与内分泌调节系统。
1.1 胃肠道不仅是重要的营养消化和吸收系统,也是重要的分子结构识别系统
人类从嗅觉到味觉,再到胃肠道和整个机体的生理生化作用、能量利用、代谢内分泌和免疫防御系统必须依赖于化学结构的识别与信号传递。这个化学结构识别和信号传递系统依赖于多个受体家族。其中最为著名的便是GPCRs超家族,它不仅有多达上千个家族成员,而且跨越光、声、电、触觉等物理信号及嗅/味觉、细胞趋化、免疫、代谢、激素和内分泌信号传递等多种化合物结构的识别与信号传递。味觉受体主要与营养、抗营养、腐败和毒性化合物识别相关联,是入“口”的第一关。由于嗅/味觉信号必须在尽可能短的时间内通过大脑作出判断,所以需通过神经电信号进行传递。已知这些信号是由GPCRs传感识别后引发胞内信号放大,迅速打开/关闭相应的离子通道,从而引发膜电位的变化(去极化),通过神经末梢收集这些电化学信号再以神经脉冲信号的形式传递到大脑。如图1所示,这种传递信号的方式也被称为非基因组学信号途径或快速信号发生与传递途径。与此同时,包括GPCRs在内的受体对分子结构的识别与传递还有一种所谓的慢速细胞信号传递途径,亦即基因组学信号途径。这条信号传递途径往往需要引发细胞内多种信号分子、接头分子的交互作用,激活(或抑制)转录因子,从而将信号传递到细胞核内,产生基因组和转录组调控作用,所以命名为基因组学信号途径。这条信号途径能够产生表观遗传修饰、细胞记忆和遗传,即在DNA和核小体上“写入(write)”“读取(read)”或“擦除(erase)”,从而改变细胞遗传记忆,而且读、写、擦方面的错误是造成疾病,特别是癌症的重要原因[15]。显然,味觉受体识别配体后可以依据其化学结构引发细胞内的信号传递,打开/关闭离子通道,引起细胞瞬时膜电位的变化,转变为神经信号传递到大脑,从而决定是否进食。但是奇怪的是,嗅/味觉受体也出现在其他组织和细胞,特别是肠黏膜组织上分布着几乎所有的味觉受体。这些受体显然不是用于向大脑传递味觉神经信号。近年来,大量研究证明,这些受体传递的是代谢、免疫和内分泌信号[16]。换言之,味蕾主要传感对食品营养和安全的信号,而肠道则主要传感植物神经和内分泌信号到大脑、脑垂体、下丘脑、胰腺、内分泌和免疫防御系统,从而控制营养吸收、转运、代谢、储存和食欲,以保证在达到其消化和吸收能力上限时停止进食。可见,营养传感、吸收与控制不仅是动物的基本属性,植物和微生物也不例外,只有在细胞需要营养时才会吸收营养,失去对营养吸收的控制也就失去了细胞生存的基础。基于此,表明这些受体是通过离子通道和耗能反应实现对营养摄入和吸收的定量化控制。以脂肪酸受体GPR120为例,饥饿时,GPR120识别脂肪酸向大脑传感“香味”,但是当肠道中传感到游离脂肪酸时,说明饮食的脂肪酸已经超过肠道的吸收能力,它转而向大脑传递“油腻”的味道,停止进食,失去这种受体会导致肥胖[17]。

图1 非基因组学和基因组学雌激素信号途径Fig.1 Nongenomic and genomic estrogen signaling pathways
植物化学物、中草药等首选口服给药方式,说明它们大多数都与肠道受体系统互作,即使可以吸收到循环系统中也必须能够很快降解并排出体外。到目前为止,超过40%的临床药物都是以GPCRs为靶标筛选得到。然而遗憾的是,肠道中到底有多少受体,这些受体的功能是什么,尚缺少系统研究,更多的食品功能评价依赖于实验动物和体外细胞实验[14]。
内源性雌激素中的17β-雌二醇(estradiol,E2)是已知3 种受体ERs(Erα、ERβ和GPER)的非选择性激活因子。17β-E2激活ERs,使之二聚体化并结合到靶基因的启动子上。另一条途径则是激活的ERs和一系列转录因子互作,形成互作网络。质膜上的ERs亚类由E2与接头蛋白(adaptor)和c-类固醇受体共激活因子(steroid receptor coactivator,Src)等信号分子互作,通过磷脂酰肌醇3激酶-蛋白激酶B(phosphatidylinositide 3 kinases-protein kinase B,PI3K-Akt)和丝裂原活化蛋白激酶(mitogenactivated protein kinase,MAPK)途径介导非基因组学快速途径。选择性激活剂E2(如G-1)或选择性ERs下调物(如氟维司群)或选择性ERs调节物(如三氧苯胺),也可以激活定位于细胞膜或内质网膜上的GPER。GPER激活腺苷环化酶合成环化腺苷一磷酸(cyclic adenosine monophosphate,cAMP),激活钙的动员和c-Src,进一步激活基质金属蛋白酶(matrix metalloproteinase,MMPs)。MMPs切割肝素结合表皮生长因子前体(pro heparin-binding-epidermal growth factor,proHB-EGF),释放游离的HB-EGF,从而反式激活表皮生长因子受体(epidermal growth factor receptor,EGFR),进一步激活MAPK和PI3K-Akt诱导外加的非基因组学快速途径,或基因组效应调节基因表达。E2介导的转录调节涉及ERs或转录因子的磷酸化,也可以直接作用于ERs,或间接结合于ERs的靶基因上。
1.2 GPER在机体通讯系统中发挥关键作用
虽然GPCRs的一个典型特征是七跨膜结构,但GPCRs的根本特征是和G蛋白相偶联,利用G蛋白上的鸟苷三磷酸(guanosine triphosphate,GTP)将其水解为鸟苷二磷酸(guanosine diphosphate,GDP)所释放的能量发挥胞内信号放大和传递作用。典型的G蛋白是由3 个亚基(α、β和γ)组成的异源三聚体蛋白。没有活性的三聚体可以通过GTP水解为GDP所提供的能量激活并解聚,分别激活下游的信号放大系统。用于激活G蛋白的能量提供者GTP一旦水解为GDP便失去再激活G蛋白的能力,需要由GTP-GDP交换蛋白(酶)换上GTP才能进行下一轮激活过程。但若要保持G蛋白的GTP供能,还需要不断把GDP再磷酸转化为GTP。由于氧化磷酸化依赖于腺苷三磷酸(adenosine triphosphate,ATP)合成酶复合物,只能提供ATP,所以GTP的合成主要依赖于α-酮戊二酸脱氢酶(α-ketoglutarate dehydrogenase,KGDH),KGDH对线粒体氧化还原状态敏感,是三羧酸(tricarboxylic acid,TCA)循环中的关键性调节酶,能够催化合成琥珀酰辅酶A和ATP与GTP之间的转换。Jesinkey等[18]报道,线粒体GTP(mitochondrial GTP,mtGTP)具有综合传感营养状态的作用,且mtGTP可以据此调节胰岛β细胞及其分泌胰岛素的功能;其经过系统研究发现:1)mtGTP可以提高钙浓度,而且不依赖于氧化磷酸化促进胰岛素分泌;2)mtGTP循环赋予的胰岛素分泌、营养传感和线粒体增大,不仅有益于健康、增加胰岛素储量,而且不会造成胰岛β细胞的“过度疲劳”;3)mtGTP配合胰岛β细胞分泌胰岛素及其代谢机制实际上构成了葡萄糖和氨基酸体内代谢平衡的关键;4)胰岛β细胞应用mtGTP传感营养的关键机制是线粒体GTP循环。在此循环中,mtGTP由琥珀酰辅酶A合成酶形成琥珀酰辅酶A,然后再在琥珀酸硫激酶的作用下,将高能硫酯键转移至GDP生成GTP和琥珀酸。这是线粒体TCA循环中唯一一个不依赖于氧化磷酸化的底物水平磷酸化。与胞浆中底物水平磷酸化不同的是,胞浆中底物水平磷酸化形成的是ATP,完全依赖于糖代谢;而TCA循环则不仅依赖于糖酵解所产生的丙酮酸经脱氢、脱羧和转乙酰基形成乙酰辅酶A,也依赖于脂肪酸β-氧化所产生的乙酰辅酶A,还依赖于回补途径所产生的丙酮酸、草酰乙酸和α-酮戊二酸,因此与糖代谢、脂肪酸代谢和氨基酸、核苷酸代谢途径构成连接和调控。mtGTP产生于TCA循环,连接并控制几乎所有宏营养代谢途径(中心代谢途径),mtGTP必须进入胞浆,通过控制G蛋白及其偶联受体发挥营养传感和吸收控制作用。mtGTP转出线粒体进入胞浆是通过线粒体磷酸烯醇式丙酮酸羧激酶,将mtGTP的高能键转移并生成磷酸烯醇式丙酮酸盐(一种高能代谢物)进入胞浆,从而整合TCA循环和回补途径信息控制葡萄糖刺激的胰岛素分泌。这一过程中的一系列调节,包括异源三聚体G蛋白、小G蛋白、线粒体GTP/GDP交换器、ATP和GTP特异性琥珀酰辅酶A合成酶异构体,都依赖于mtGTP循环。Jesinkey等[18]研究还证明,mtGTP在体外和体内诱发并放大葡萄糖刺激的胰岛素分泌,增加mtGTP的合成,增加钙离子在葡萄糖刺激下的胰岛素分泌幅度;mtGTP还可以增加线粒体质量、胰岛素颗粒体数量和不引发脱分化的膜接近尺度或代谢脆性,强调了mtGTP信号在营养或激素传感、胰岛素分泌、线粒体维护和胰岛β细胞健康等方面的重要作用。线粒体中的草酰乙酸盐、琥珀酸盐、线粒体磷酸烯醇式丙酮酸羧激酶通量都会增加mtGTP循环。回补的丙酸盐可以通过丙酰辅酶Aβ变构机制激活丙酮酸羧化酶,在高浓度下抑制胰岛素分泌[19-20],其原因是丙酸盐减少基础琥珀酰辅酶A合成GTP。总之,虽然GTP/GDP和ATP/ADP之间可以互相转化,但是它们只在核苷酸合成,特别是DNA复制原料准备时发挥作用,而在营养、激素和氧等传感中发挥关键作用时存在明显而严格的分工,因此,只有GTP才可以作为G蛋白信号放大的直接驱动力,而ATP则只能通过磷酸化靶蛋白或化合物发挥激活作用。
细胞对环境变化,特别是环境中营养的传感与应答都必须通过快、慢两条信号途径,前者是通过非基因组学途径控制离子通道开/关;而后者则依赖于基因组学信号途径传入细胞核。这两条途径都依赖于异三聚体G蛋白或小G蛋白,也就是依赖于GTP循环。而ATP则主要通过各种激酶激活胞内信号途径。GTP不仅为异三聚体G蛋白提供动力,也为小G蛋白提供动力,蛋白质和RNA的核内、外转运,蛋白质生物合成的起始、延伸等都依赖于GTP,特别是当细胞(包括原核细胞)在碳源和氮源饥饿时,更要依赖于GTP和ATP合成警告素(alarmone)传递预警信号,如通过空载的tRNA进入核糖体A位所传递的氨基酸缺乏信号(guanosine 5’-diphosphate 3’-diphosphate,(p)ppGpp),也就是通过激活Relaxed A(RelA)和Spotless T(SpoT)或RelA/SpoT同源蛋白RSH合成。RelA是一种核糖体相关(p)ppGpp合成酶,负责将ATP的焦磷酸转移到GTP的3’-羟基上,从而合成(p)ppGpp,之后直接以GTP为底物合成ppGpp[21]。SpoT是一种ppGpp合成/水解双功能酶,在生理上与酰基载体蛋白合作,所以它也是一种脂肪酸代谢辅助因子[22]。显然,这种互作也是脂肪酸饥饿促进合成ppGpp所必需的。SpoT也合成ppGpp,对环境作出响应,包括磷、碳源饥饿。RelA/SpoT同源蛋白几乎在所有细菌中都可以检测到,说明ppGpp依赖性严谨反应是一个通用的通过调节自身代谢途径、细胞分裂、转录和翻译控制适应营养源和环境条件的应激系统[23]。大量研究表明,ppGpp不仅是氨基酸类氮饥饿传感和应激系统,而且是磷饥饿[24]、碳饥饿、脂肪酸饥饿、各种毒素[25]、抗生素及其耐受[26-27]、干旱、高渗和低渗、高温、低温[28]、氧化[29]等恶劣环境的应激响应系统[30]。值得注意的是,近年来发现广泛分布于所有原核细胞的这一应激系统也存在于真核细胞和高等动、植物中。
Sun Dawei等[31]在后生动物中鉴定到SpoT同源序列并命名为Mesh1,人类由HDDC3基因编码,果蝇则由Q9VAM9基因编码。他们详细研究了这些蛋白的结构与功能发现,像细菌的此类酶一样,Mesh1蛋白含有一个水解ppGpp的活性位点和一个保守的His-Asp-box镁离子结合基序。与这些结构相一致,Mesh1可以在体内、外有效地水解ppGpp的3’-和5’-磷酸。Mesh1也可以抑制SpoT缺陷型致死和RelA诱导的细菌细胞生长延迟。果蝇Mesh1(Q9VAM9)基因缺陷也可以造成生长迟缓和抗饥饿能力受损。微阵列芯片分析表明,在氨基酸饥饿时,Mesh1基因缺失突变将强烈下调DNA和蛋白质合成相关基因表达,并上调应激响应基因表达。证明后生动物SpoT同源序列具有饥饿响应的保守功能[32]。换言之,除有些内源性激素可以通过受体酪氨酸激酶向细胞内传递信号外,几乎所有外源信号和趋化因子等内源信号都需要通过受体和G蛋白偶联向细胞内传递和放大信号。
2 GPER-ER-AHR-EGFR信号调节网络
核受体作为经典的配体激活转录因子介导需要较长时间的基因组学信号途径。现在已经基本确定固醇类配体也可以通过GPCRs介导的快速响应途径,也就是非基因组学途径[32]。虽然已有研究发现经典的固醇类配体能够直接激活经典固醇受体传递快速信号,但是越来越多的证据表明,这些作用是因为转膜受体的存在。其中最重要的受体就是GPER,它作为一种GPCR,介导雌激素依赖激酶激活,从而发挥快速响应和转录应答作用。这些响应通常分为两类:1)基因组学响应,其特点是需经过改变转录的信号传递过程,往往需要数小时甚至数天的时间;2)快速信号途径,即刺激后即刻作出响应,只需几分或几秒的时间。前者通常与配体依赖性转录因子激活相联系,而后者则与生长因子受体和GPCRs相联系,通过打开或关闭离子通道,如钙离子通道,传递信号。GPCRs激活方式也涉及到激酶的激活和NO等第二信使的信号传递过程。这些快速信号传递属于非基因组学应答途径,有别于经过转录控制的信号途径。然而,这些区别仍然依据人为标准评判,因为所有激素(固醇类或其他)似乎都是通过受体协同控制引发快速和慢速两个信号途径。慢速途径,即经典途径,往往涉及转录应答过程。而且,非基因组学途径同时也引发慢速的基因组学途径,这两个途径之间通过复杂的机制互相协同,进行快速和慢速两个水平上的协调与控制。
2.1 经典固醇受体
固醇类激素家族包括:黄体酮、睾丸酮、糖皮质激素、盐皮质激素,其中雌激素是研究最为透彻的成员。固醇类配体的高疏水性使它们可以通过被动扩散进入细胞膜[33],但是这同时也使得它们更可能聚集在细胞膜上[34]。此外,关于雌激素基于固醇基团E2、雌激素酮和雌激素酮硫酸盐的生物调节功能的研究已有很多[35]。雌激素活性物质包括天然和人工合成的多种化合物。天然雌激素类化合物多发现于植物,即植物化学物或植物雌激素。典型的植物雌激素类化合物是黄酮类,如香豆雌酚、异戊二烯化黄酮类或异黄酮[36]。人工合成的雌激素类化合物,即外源性雌激素、环境雌激素和内分泌干扰物,包括各种杀虫剂、多氯联苯二酚、增塑剂以及无处不在的现代社会所暴露的各种低浓度污染物[37]。这些植物和外源性雌激素的主要生理作用是通过经典的ERs途径[38]发挥类似雌激素的作用。虽然称之为雌激素,但是也在正常的男(雄)性生殖中发挥关键作用。至少有两种典型ERs在人类和啮齿类动物睾丸中都有表达,但是这些受体在不同细胞中的表达模式和产物有所不同[39]。无论芳香酶或ERα基因删除都会导致精子发生混乱和不孕。但是两种激素的动物模型表现出明显的生理、细胞和内分泌水平上的不同[40-41]。在雌性动物中,删除ERs基因将会失去雌激素依赖性信号途径。芳香酶缺陷和ERα缺陷小鼠相互交叉的表型本质揭示了可能还有其他ERs也发挥重要作用。雌激素依赖信号途径也在很多非生殖系统中发挥关键性保护作用,包括心脑血管、骨骼和神经系统。有研究表明,雌激素在多个水平上对心脑血管疾病具有保护作用,包括心脏肥大、收缩力减弱、血管柔韧性和胆固醇代谢等[42]。强有力的证据显示,多种ERs参与了基因组学和非基因组学信号传递过程。与此相似,雌激素对骨骼和神经系统的保护功能相当复杂,涉及多种受体和信号途径[43]。所有这些作用都涉及到ERα和ERβ,而且还涉及到GPER,它们可能通过调控基因组学和非基因组学两条信号途径[44]发挥作用。最早描述的ERs(先定名ER,后定名ERα)是1973年在大鼠的子宫/阴道中提取得到的[45],其DNA序列测定于1986年[46],其配体结合结构域的晶体结构则报道于1997年[47]。第2个ERs,ERβ鉴定于1996年[48],其受体-配体作用及其核转录因子调节机制是识别激素相关基因启动子的顺式激活激素应答元件。虽然ERβ和ERα在DNA序列上具有高度同源性,但是在配体结合结构域、转录激活作用结构域的同源性却相对缺少[49],这表明它们属于功能上相互区别的两个不同亚类,这也从小鼠敲除ERα和ERβ的研究中得到了进一步证明[50]。ERs先是存在于细胞核中,在细胞核内和伴侣蛋白(chaperones)形成复合物。当配体结合时,其构象改变,导致伴侣蛋白解离,受体二聚体化,然后在相应的启动子位点和DNA结合,招募辅助因子和/或辅助抑制因子,导致基因表达速率的改变。
2.2 固醇类激素通过GPER受体传递信号
已知雌激素和其他固醇类物质都传递长时间的转录调节信号,但是所有这些信号都是以快速信号传递作为起点[51]。对于快速信号传递,早在1967年有关固醇类激素发挥转录活性的问题一直存有争议[52],因为从逻辑上分析,转录活性不可能在没有信号传递的情况下发生。例如,经典的固醇类受体可以在配体的直接刺激下结合到DNA上发挥转录调节作用,也可以在没有配体存在的情况下发挥激活作用[53],因为它们可以直接结合到DNA上并不依赖于骨架转录因子,如激活蛋白1(activator protein-1,AP-1)[54]。而且,经典的固醇受体也可以介导激酶的激活作用,如MAPKs、PI3Ks和Src,从而通过磷酸化作用导致转录因子,如Elk-1[55]和血清反应因子(serum response factor,SRF)的激活[56]。由固醇类或其他受体类型所产生的激酶的激活作用也可以产生固醇类受体的磷酸化,导致转录激活[57]。因此,快速激活作用可能是经典固醇类激素受体细胞信号传递的重要效应。尽管经典的固醇类受体细胞应答和激活作用的很多机制都得到了阐述,但是固醇所介导的生理效应在很多系统,包括神经系统、免疫系统、胰腺和骨骼系统的作用机制尚未完全明确[58]。尽管在很多情况下,这些效应被描述为经典ERs的膜相关形式所介导[59],但是一系列经典的ERs(特别是ERα)敲除小鼠实验研究证明,可能还有其他受体发挥作用[60]。例如,应用不能透过细胞膜的雌激素,同样可以激活雌激素膜受体[61],应用ER拮抗剂也可以得出同样的结果,而且该受体需要G蛋白的参与[62]。另外,ERα敲除小鼠仍然表达ERβ,即使ERα/ERβ双敲除小鼠仍然可以表达选择性剪切的ER形式[63],或者通过补偿的发育途径使不存在此途径的鼠科动物生存。Filardo等[64]于2000年首先鉴定到这样的受体,并证明该受体为GPR30/GPER,涉及缺少经典核受体ERs的雌激素介导的细胞外调节蛋白激酶(extracellular regulated protein kinases,ERK)1/2-MAPK激活作用[64]。在MCF-7细胞系中,雌激素介导快速增加ERK1/2的磷酸化,表达ERα、ERβ和GPER,但是在MDA-MB-231细胞系中,则表达ERβ,但不表达ERα或GPER,这种应答缺失说明ERα[65]或GPER发挥重要作用。将GPER转染到MDAMB-231细胞系即可恢复对雌激素的应答。这一信号传递应答也存在于KBr3细胞系,该细胞系表达GPER但缺少ERα和ERβ。总之,这些结果进一步强调GPER的确参与了雌激素的信号传递。进一步的研究还表明,GPER的激活作用还涉及EGFRs的反式激活机制。GPCRs对EGFR的反式激活途径相对来说还是一个新的概念,涉及Shc蛋白、生长因子结合蛋白和金属蛋白酶对proHB-EGF的切割作用[66]。Filardo等[67]描述了GPER介导的cAMP升高可以通过表皮生长因子(epidermal growth factor,EGF)激活ERK1/2使雌激素恢复到基础水平,其作用机制依赖于蛋白激酶A-Raf-1的激活或抑制[67]。因此,这些核受体显然受各种磷酸肌醇磷酸盐的激活,通过GPER和经典的ERs接受雌激素刺激,从而提供了一种精致的固醇类激素介导的不依赖于传统的固醇应答元件的转录控制机制。对经典ERs拮抗剂、激活剂的广泛研究和鉴定,进一步促进了GPER生物学作用的阐明。应用虚拟/分子生物学筛选方法鉴定和GPER的选择性,Bologa等[68]的研究结果表明,G-1可以抑制E2-Alexa结合是通过在表达GPER细胞中竞争性结合雌激素到E2-Alexa,其抑制常数Ki为5.7 nmol/L,而G-1的Ki仅为11 nmol/L。对ERα和ERβ表达细胞系的类似操作所得到的雌激素Ki分别为0.30 nmol/L和0.38 nmol/L,即当G-1浓度达到1 μmol/L时,没有显著性结合。GPER转染COS7细胞系的结果显示,G-1是一种激活剂。为了证明G-1对GPER的选择性作用,与ERs相比,COS7细胞系不管表达ERα还是ERβ都对G-1没有响应,但是对ERα和ERβ产生了快速响应。G-1也可以激活内源性GPER,并导致PI3K的激活。
雌激素可以获准自由进入细胞,从而与胞内ERs(包括ERα、ERβ和GPER)结合。在细胞中,GPER或ER表达在细胞表面,所以雌激素并不需要跨过细胞质膜。雌激素结合到经典的ERs上,导致直接的转录激活和信号传递。当激活剂结合到GPER时,可以激活异源三聚体G蛋白,然后通过G蛋白激活多种效应因子,包括腺苷环化酶催化cAMP的合成、Src和鞘氨醇激酶。之后涉及2 条途径:激活MMPs切割proHB-EGF,释放游离HB-EGF;或反式激活EGFR。EGFR的激活导致多种下游事件的发生,包括磷酸酯酶C(phospholipase C,PLC)、MAPKs和PI3Ks的激活。PLC的激活再通过肌醇三磷酸(inositol triphosphate,IP3)控制胞内钙储藏和钙离子通道开关。与此同时,MAPKs和PI3Ks的激活导致一系列胞浆途径的激活,包括核蛋白和转录因子的调节作用。因此,雌激素刺激可以升高其启动子不含固醇应答元件的靶基因表达。这种联合效应是通过胞浆信号和核转录事件共同作用于细胞增殖的结果。

图2 雌激素介导的GPR30信号传递机制[32]Fig.2 Estrogen mediated GPR30 signaling mechanism[32]
雌激素通过经典的ERs和GPER共同介导快速和基因组学信号传递及其交谈,如图2所示。雌激素可以直接激活经典的ERs,从而经辅助激活因子和辅助抑制因子直接调节转录活性。ERs和GPER两种受体可以选择性激活多种激酶途径,导致ERs的磷酸化;或者通过选择辅助调控因子与ERs互作。MAPKs和PI3K也会产生转录因子的磷酸化激活作用,如Elk-1和SRF。由PI3K催化所产生的磷脂酰肌醇三磷酸也可以导致其他核受体家族成员的激活,如固醇合成因子1、肝受体类似物1和cAMP都可以升高并激活转录因子,从而调节cAMP应答元件(cyclic AMP response element,CRE)结合蛋白和转录活性。ER和GPER似乎也受到调节过程的控制,当配体结合到ER时,同时也启动了最终的蛋白降解;而多数GPCRs也都服从脱敏机制。几乎所有信号途径都显示出潜在的互作特性,无论正的还是负的,导致信号传递和调节的极大复杂性;而多种表型输出又都依赖于细胞中各种信号分子之间复杂的相互作用。雌激素、其他固醇类受体、配体、生长因子和GPCRs都可能反馈信号到信号途径,在受体和靶基因(CRE、雌激素应答原件(estrogen response element,ERE)和血清应答原件)表达调控之间形成一个更加复杂的细胞活动“交响曲”。
EGF受体受GPCR的反式激活作用,并需要金属蛋白酶切割proHB-EGF。Prenzel等[69]等研究发现,proHB-EGF和金属蛋白酶活性是GPCR信号传递和EGFR激活的关键。GPCR诱导的EGFR反式激活机制代表了一种新的认知,因为这需要3 个膜信号传递事件,即配体和GPCR互作激活异源三聚体G蛋白,导致一个细胞内的信号产生,这个信号进一步诱导跨膜金属蛋白酶激活,从而诱导膜生长因子前体的胞外加工,从而释放成熟的因子,然后直接或通过基质蛋白聚糖和EGFR胞外结构域互作,最后激活胞内信号传递,如图2所示。这种新机制为前列腺癌细胞病理生理学提供了一个合理的解释,因此他们假设[64],EGFR通过G蛋白介导的生长因子前体酶解激活,代表了一个广泛的反式激活新机制。并且,一系列具有多样性生物活性多肽,如肿瘤坏死因子α(tumor necrosis factor α,TNF-α)、FAS-配体或L-选择素,都是膜蛋白前体28的加工产物,它们与多种生理紊乱相关,所以这些膜相关蛋白酶的加工和成熟可能成为疾病治疗的重要靶标。
3 核受体超家族及其调节网络
核受体超家族由500多个成员构成,该家族依据其主要特点,如二聚体化、DNA结合基序、特异性以及配体结合情况进一步分为4 类:固醇类受体为第1类、视黄醇X受体异二聚体为第2类、同型二聚体孤儿受体为第3类、单体孤儿受体为第4类[70]。虽然有一些重要的结构与功能出现在两类之间,但部分关键结构是保守的,可以预测功能,仍然维持着4 类的分类系统[71]。所有核受体超家族成员都含有一个可变的N-末端结构域(N-terminal domain,NTD)、一个DNA结合结构域(DNA binding domain,DBD)、一个铰链区、一个保守的配体结合结构域(ligand-binding domain,LBD)和1 个可变的C-末端结构域(图3)。2 个最为保守的结构域构成核受体的DNA结合结构域和配体结合结构域。DNA结合结构域含有2 个锌指基序,用其钩住,并结合到细胞核中的染色质上[72]。每一类核受体都具有2 个DNA结合识别序列,分布于两边的可变位点,带有反向重复、直接重复或不重复的DNA序列[70]。核受体的配体结合结构域保持功能上的高度保守性、双向特异性和特异性配体的亲和性[73]。除孤儿受体外,所有受体都由配体激活。配体结合到配体结合结构域,诱导变构作用,继而激活受体[73]。每一类核受体的配体都具有相似的结构,并且配体的分类决定了核受体的类属[73]。例如,内源性表达配体所作用的受体往往都是激素、酶催化配体或尚未确定的配体相关受体[73]。
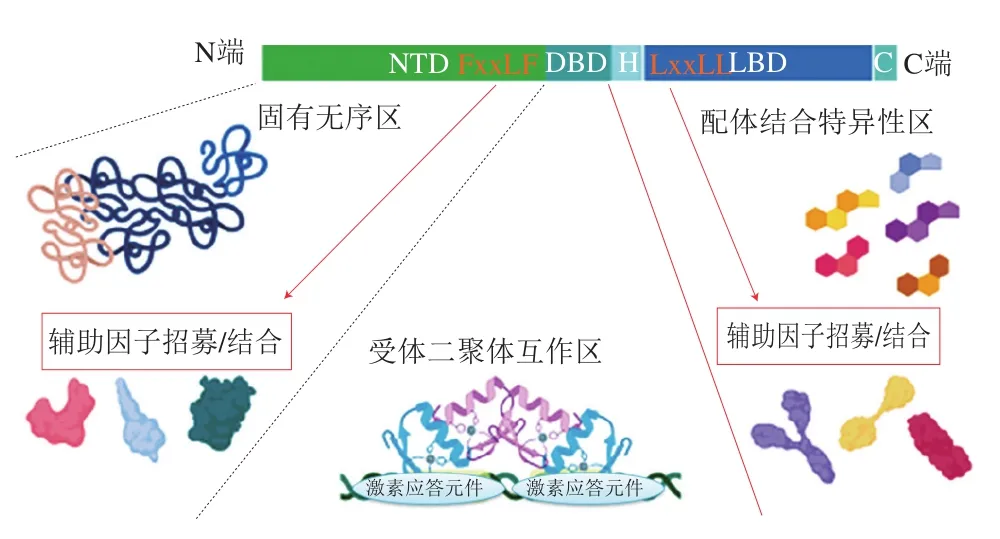
图3 激素家族核受体示意图[70]Fig.3 Schematic diagram of the hormone nuclear receptor family[70]
雌激素-受体调节转录在乳腺癌细胞中涉及蛋白辅因子对染色质结构的调控。这个过程包括辅因子对组蛋白的修饰,如乙酰化和甲基化,因此改变靶基因的转录状态。已经有大量特异性辅因子与ERs互作并招募到特异性启动子区域的研究,Green等[74]对这些作用进行了综述,系统分析了这些雌激素-受体互作,招募辅因子对染色质进行修饰,改变靶基因的转录状态及其在乳腺癌发生、发展和转移中的作用。
3.1 ERs的结构
和其他激素应答核受体相似,ERα和ERβ也包含几个保守的功能结构域。一个N-末端转录激活结构域AF-1位于A区和B区,该区在ERα和ERβ之间存在明显变异;一个DBD定位于C区内,由66 个氨基酸残基构成,包含2 个非等价的富含半胱氨酸的锌指[75-77]。N-末端的锌指结构域负责识别ERE,序列为GGTCANNNTGACC[78],第2个锌指结构负责稳定非特异性蛋白和DNA的互作。锌指结构还具有约束ER,防止与非经典或不完美的ERE DNA基序相结合的作用[79]。D区是铰链区,E区作为配体结合区,属于LBD,并作为第2个激活结构域AF-2的基础。与之相邻的是F区,是一个可变结构域。整个ER的很多不同结构域都可以通过与特异性辅因子互作发挥不同功能,但很清楚的一点是,ER的AF-1和AF-2结构域是染色质模板激活发挥有效活性所必需的[80]。ERs的LBD三维结构相似于核受体超家族[81],雌激素和三苯氧胺(他莫西芬)都是结合在ER的同一个LBD螺旋形口袋中[47,82]。ER的螺旋3、5和12定位于AF-2结构域,负责识别并连接蛋白组分,组装转录装置,这些螺旋的变异会特异性消除AF-2的活性,导致配体依赖性激活作用。当ER结合雌激素时,在AF-2结构域与辅因子p160蛋白家族基序LxxLL互作。p160蛋白家族由3 个成员:SRC1、谷氨酸受体相互作用蛋白1(glutamate receptor interacting proteins 1,GRIP1)和乳腺癌扩增性抗原1(amplified in breast cancer 1,AIB1)构成,是ER辅因子中最有特色的成员。SRC1的LxxLL基序与ER的AF-2结构域之间的互作发生在短的两亲性螺旋和ER表面螺旋3、4、5和12所形成的互补大沟之间[83]。这种构型上的变化使ER蛋白和其他辅因子之间能够正确互作并定位,从而促进靶基因转录。当三苯氧胺的活性代谢物4-羟基三苯氧胺结合到ER的LBD时,就会诱导其螺旋LxxLL基序模拟辅助激活因子螺旋产生螺旋12结构重排,并因此阻遏p160辅助激活因子的结合。然而,三苯氧胺的存在不仅可以通过防止辅助激活因子结合到ER上从而抑制转录,而且可以改变ER的结构,从而优先与第二类辅因子,辅助抑制因子(包括核受体共抑制因子1(nuclear receptor corepressor 1,NCOR1)和核受体共抑制因子2(silencing mediator of retinoid and thyroid hormone receptors,SMRT))[84-86]互作。雌激素和三苯氧胺两者结合则具有改变ER三维结构形成、打开或关闭嵌入位点状态的作用,形成一个“闸刀”开关。
3.2 ERα与辅因子互作
Kos等[63]用染色质免疫沉淀法研究ERα与辅因子互作,结果显示,在体内ER和p160辅因子可以进、出组织蛋白酶D(cathepsin D,CTSD)和三叶因子1(trefoil factor family 1,TFF1)启动子(Box2)循环,并可以预计其转录方式[87-88]。ER和p160控制相关基因启动子循环的生物学意义尚不完全清楚,但理论上是控制染色质结构的疏松和开放,以保持多次转录与循环。当然这似乎是一个过分简单的理解,也许这种多次转录循环还需要持续的染色质调控,以便保持常染色质条件下有足够的时间组装转录装置,保证其后的多次基因表达循环。因此,辅因子的作用可以直接调控染色质,招募蛋白因子,调节染色质重构属性才是最关键的。用三苯氧胺处理的结果显示,伴随雌激素-ER互作,ERα被招募到TFF1启动子上,控制TFF1启动子出、入循环动力学状态。然而,取代p160辅助激活因子的招募,辅助抑制因子NCOR1和SMRT则参与了抑制过程,表明辅助抑制因子调控染色质可能是通过基因表达抑制中的重要控制元件。一系列的证据支持辅因子在化学计量学水平上的波动控制了ERα转录活性以及平衡和循环变更机制。AIB1的水平在肿瘤中增加50%以上,在乳腺癌中约放大10%[89],这表明AIB1的确在乳腺癌发生和发展中发挥作用。最近的体外研究结果也证实,NCOR1和SMRT可以直接发挥抑制作用,负调控三苯氧胺的抗肿瘤细胞增殖的治疗作用[90]。上述这些研究都强化了不同ER辅因子在乳腺癌发生、发展和治疗中具有不同的基因表达和调控作用。
3.3 ER的转录辅因子与酶活性
近来,最有意义的贡献就是对转录控制机制的深入研究,而这些研究是对组蛋白的修饰和鉴定所带来的。因为组蛋白的修饰控制着染色质是否处于转录因子易接近状态和转录机制。靶基因的转录激活作用需要染色质在转录起始位点解凝聚,才能促进转录起始过程。组蛋白N-末端加上乙酰基是通过组蛋白乙酰基转移酶(histone acetyl transferases,HATs),而脱掉乙酰基则通过组蛋白脱乙酰基酶(histone deactylases,HDACs)催化[91]。乙酰化作用在赖氨酸残基上特异性发生,从而导致赖氨酸正电荷被中和,弱化了组蛋白上赖氨酸侧链与DNA互作[92],增加了转录装置对染色质的易接近性[93]。如上所述,雌激素结合ERα即可招募p160蛋白SRC1、GRIP1和AIB1,所有被招募的蛋白都有一个基本的螺旋-环-螺旋基序,用来介导蛋白-蛋白互作[94]。p160蛋白家族已显示出在雌激素存在时招募HATs到ER-转录复合物的作用。所有p160蛋白都可以和CREB结合蛋白(cAMP response element binding protein binding protein,CBP)以及p300辅助激活因子互作[95],具有内在的HAT和激活组蛋白亚基3的14位赖氨酸(H3K14)、H4K5、H4K8乙酰化活性,拮抗H2A和2B激活[96],以及组蛋白H3的14位赖氨酸激活[97]。除和p160辅助激活因子互作之外,p300也可以直接与ER的AF-2结构域互作[98],并通过与AF-1和AF-2互作增强其协同作用,促进TATA结合蛋白(TATA binding protein,TBP)结合[99-100]。尽管如此,p300和CBP在生物活性方面很多重合,例如,两者对雌激素靶基因启动子都有周期性,但是又有所不同。一个可能的解释是,p300和CBP两者都是TFF1启动子染色质初期解凝聚所必需的[101],在染色质还没有完全再凝聚时,又开始了新一轮的转录,但是新一轮转录依赖于CBP保留在常染色区。在前列腺癌细胞系中,p300可以直接调节雄激素受体的乙酰化状态[102],并可作为靶基因染色质上的雄激素受体辅因子[103],这表明HATs对激素抵抗具有调节作用。另一个具有HAT活性的ER相关因子是p300/CBP相关因子(p300/CBP-associated factor,PCAF)[104],这也是酵母菌辅激活因子GCN5的同源蛋白[105]。PCAF与ER互作是间接的,PCAF可以自乙酰化或被p300乙酰化,从而提供反馈调节回路[106]。PCAF对染色质的结构变化也有作用,通过乙酰化H3K9和H3K14增加基因转录。大多数相关研究工作集中在感兴趣的1~2 个启动子区,但是后来的研究表明,启动子的乙酰化一般都发生在ERα介导的基因转录期间[107]。生长因子和ERα之间的互作已经得到了较充分的研究,并且与非基因组学途径紧密相连。但是,ERα更复杂的情况可能与激酶有关。雌激素还可以诱导ERα和核因子κB激酶、核因子κB抑制因子激酶(inhibitory nuclear factor κB kinase α,IKKα),在CCND1启动子上互作,其结果导致IKKα介导的组蛋白H3[108]诱导CCND1转录及其磷酸化。周期蛋白D1单独足以诱导乳腺癌细胞起始DNA的合成[109],IKKα可以调节周期蛋白D1,证明它们也在细胞周期调控中发挥非常重要的作用。
3.4 三苯氧胺-ER辅阻遏因子
很多基因,特别是控制营养吸收和生理代谢过程的基因在不需要继续转录时,就会关闭。而抑制(关闭)这些基因的转录需要将乙酰基团从组蛋白尾巴上去除,导致染色质凝聚。三苯氧胺结合ER,可以进一步招募辅阻遏因子NCOR1和SMRT1,这一功能主要用作接头分子,进一步招募并控制HDAC的激活作用。ER-三苯氧胺复合物通过被称为CoRNR框(CoRNR boxes)的基序与ERα的螺旋3和5互作,这些都是构成招募ERα转录抑制的关键。NCOR1和SMRT与ER-三苯氧胺复合物互作,进一步导致与HDAC3、HDAC1和核小体重组装以及组蛋白脱乙酰化复合物Nu-RD在TFF1启动子上的互作[110]。三苯氧胺结合NCOR1-HDAC3和Nu-RD-ER-复合物是有区别的,虽然两者都可以结合在相同的启动子区域,但是它们并不是同时出现,表明可能是一个快速交换不同辅阻遏因子复合物的过程[110],以保证处于异染色质的环境。
ERα开启和关闭形成约45 min的染色质转录循环。这些循环由以下系列事件组成:先驱因子FOXA1结合到异染色质上(图4中I部分所示)决定ERα结合的基因组,然后进行雌激素刺激,ERα与TFF1启动子联合,然后HATs被招募进行局部乙酰化(图4中II部分所示)。与此同时,组蛋白甲基转移酶(histone methyltransferases,HMTs)结合,p160辅助激活因子也在TFF1启动子上聚集。RNA聚合酶II(RNA polymerase II,PolII)和转录装置在ERα后面加载并伴随组蛋白甲基化增加(图4中III部分所示)。当染色质上ERα蛋白减少时,剩下的ERα与SWI/SNF复合物的各组分联合,HATs和AIB1以及转录装置处于低水平(图4中IV部分所示)。当ERα水平又开始增加时,下一轮循环伴随着HMTs、转录因子、HATs(不是p300)、适配体蛋白和p160辅因子的增加而开始(图4中V部分所示)。当ERα水平在染色质上达到最大值时,HMTs和p160蛋白又开始减少,只留下HATs、适配体蛋白和转录装置(图4中VI部分所示)[110]。

图4 染色质免疫沉淀法测定ER和辅因子在TFF1启动子所启动的循环动力学Fig.4 Cycling kinetics of ER, and co-factors at the TFF1 promoter as determined by chromatin immunoprecipitation assay
如图4所示,辅阻遏物水平变化提示其在调节乳腺癌进程中发挥作用。在乳腺癌中,NCOR1水平下降,表明癌症开始扩散[111]。在模型鼠中NCOR1水平下降意味着三苯氧胺抵抗[112]。同样,在乳腺癌患者中直接抑制NCOR1或SMRT可以逆转三苯氧胺对肿瘤生长的抑制作用[113]。雌激素可以抑制NCOR蛋白水平,并不改变其mRNA水平[114],这可能是由蛋白酶体降解NCOR所造成的[115]。此外,还发现雌激素降低NCOR1水平的证据有,低水平的NCOR1往往对应于晚期乳腺癌或乳腺癌对三苯氧胺反应迟钝,这可能是由辅阻遏物化学计量学改变影响了ERα的转录输出引起,当然这还需要进一步的临床证据。最近的研究已经获得了ERα介导抑制作用的直接证据,证明周期素G2(cyclin G2,CCNG2)基因是雌激素抑制靶基因,ERα的确可以直接招募HDAC1和NCOR1到CCNG2启动子上[116]。
3.5 核受体通过辅助因子所形成的调节网络[117]
核受体-辅助调节因子之间的互作研究开始是基于细胞为基础的实验方法[118-121]以及辅助调节因子-核受体互作微阵列实时检测技术(microarray assay for realtime coregulator-nuclear receptor interaction technology,MARCoNI),用这些方法可以比较配体与受体,包括核受体-突变体之间的互作[122-123]、核受体翻译后的修饰及其对核受体-辅助调节因子互作的影响等[124]。
在无配体的条件下,辅阻遏物通过LxxxIxxxL基序(L代表亮氨酸、I代表异亮氨酸、x代表任何氨基酸)互作停靠到AF2结构域的表面(螺旋3/4/5/12界面)[125]。当配体结合后,LBD经历一个构象上的变构作用,涉及重新定位和螺旋12的稳定化,导致辅阻遏物结合与释放,以及辅激活物(亦即辅调节开关)的招募,最后激活靶基因的过程。辅激活物通过LxxLL基序结合核受体[126]。配体的本质决定了螺旋12的精确位置以及随后与辅激活物的互作[127]。一些辅阻遏物,包括配体依赖性辅阻遏物通过LxxLL基序结合到核受体上[128]。辅阻遏物基序和辅激活物基序形成一个两亲性α-螺旋,其疏水性残基停靠在核受体的LBD界面上。核受体的LBD互作可能并不能认作所谓两相开关(“on/off” switch)模型,更应该把核受体的LBD看作“可变电阻”,调控不同核受体结构域和辅助调节因子变构之间的微调变构机制,从而产生一个分级信号输出[129]。
至少已有350 种核受体辅助调节因子得到鉴定[130]。核受体辅助调节因子形成一个结构多样的蛋白质族群,并常常表现出遍在的表达模式。辅助调节因子在对核受体特异性和亲和性上往往表现出相互重叠。这些互作常常是短暂的,含有很多辅助调节因子和多个互作基序,因而产生一个庞大的,具有不同潜力、不同程度的互作,导致核受体介导的基因表达输出[131]。核受体-辅助调控因子之间的互作体现在由核受体-辅助调节因子信号传递所引起的各种特异性疾病上。至少有160 种辅助调节因子与病理学状态有关,包括各种类型的癌症[132]。关于核受体-辅助调节因子互作,包含应用各种肽类为基础的方法、时间分辨荧光能量转移[133]、噬菌体展示技术[134]和基于多路复用微球方法配合流式细胞术[135]等所做的研究。高通量谷胱甘肽S-转移酶下拉实验[136]、基于双杂交的方法[137]和蛋白微阵列[138]都提供了一系列深入调查经纯化的全长辅助调节因子和核受体蛋白互作研究工具。但是在这些研究中,并不能为核受体的完整特性、互作的具体机制以及为核受体家族成员提供更多具体信息[139]。除一般结合作用(例如,α-螺旋的完整性在辅助调节因子基序上的完整性)外,核受体-辅助调节因子互作的特异性、选择性等仍知之甚少。
高通量核受体-辅助调节因子分析预期可以改善对核受体-辅助调节因子互作组学的了解。应用MARCoNI技术[140],Broekema等[117]系统分析了24 种核受体和154 种辅助调节因子基序之间的结合,获得了3 696 个核受体-辅助调节因子互作数据,建立了每一种核受体-辅助调节因子独特的互作数据。对核受体-辅助调节因子数据依据核受体氨基酸序列进行层次和核受体-辅助调节因子精细互作聚类分析,结果表明,该核受体-辅助调节因子肽互作组学数据分析可以为营养、临床、药物筛选、设计与应用的研究提供一个开放的数据资源。核受体和辅助调节因子之间的互作动力学在核受体介导的基因调控中发挥重要作用。通过细胞独立因果分析和基于肽的微阵列分析,Broekema等[117]还系统生成了3 696 个核受体-辅助调节因子互作整体图谱。虽然核受体亚家族表现出相似的辅助调节因子互作轮廓,但是每一种核受体都有其独特的辅助调节因子互作特性,反之亦然。辅助调节蛋白的LxxLL和LxxxIxxxL基序具有不同的核受体特异性、基序选择性,甚至混合特性。这些数据还表明,核受体的LBD和辅助调节因子基序两者的氨基酸序列都在决定辅助调节因子结合中发挥重要作用。
核受体是脊椎动物最大的转录因子家族,几乎调节着所有生理活动[141],包括细胞生长与分化、代谢进程、生殖、发育和体内平衡[142]。核受体的转录激活作用受内源亲脂性化合物和辅助激活因子或阻遏物的控制[143]。人类基因组含有48 个核受体成员[71],序列比对和系统树构建结果表明,人类核受体(human nuclear receptor,hNR)家族分为7 个进化上相同的亚家族[144]。剂量敏感的性别反转-先天性肾上腺发育不良蛋白1(dosagesensitive sex reversal- adrenal hypoplasia critical region on chromosome X protein 1,DAX1,又称NR0B1)及其紧密相关的Src同源区2蛋白酪氨酸磷酸酶(Src-homology domain 2-containing protein tyrosine phosphatase,SHP,又称NR0B2)是两种非典型孤儿受体,属于hNR家族的NR0B亚家族,它们缺少典型的锌指DBD和其他hNR成员的辅阻遏物活性[145]。这两种蛋白质在哺乳动物代谢、生殖、营养和甾醇类合成中发挥重要作用,它们的突变体往往产生多种疾病,如癌症、肾上腺发育不良、罕见的男性遗传病,并且伴随低促性腺素性功能减退症等疾病[146]。DAX1具有一个独特的N-末端结构域三重复区,三重复的每一个结构域又拥有一个保守的富含亮氨酸受体结合基序,即LxxLL基序,该基序通常发现于hNR辅助激活因子[147]。SHP也含有两个LxxLL基序,一个位于N-末端结构域,另一个位于C-末端结构域[148]。LxxLL基序通过特异性识别并与AF-2结构域互作介导DAX1/SHP的同或异二聚体与各种hNR成员互作[149]。最近,在人类基因组中鉴定了一系列DAX1/SHP缺陷症,包括NUR77、组成型雄烷受体(constitutive androstane receptor,CAR)、SF1和LRH1;它们涉及多种生理功能和病理进程,例如,异生物质代谢、亚细胞定位、原发无精症和先天性肾上腺发育不全[150]。于是,Wu Tao等[151]成功设计了一系列可以驱使构象改变,瓦解癌基因转录辅阻遏物与NaC1及其伙伴POZ蛋白形成二聚体的构象。Walensky等[152]同时还设计了一种碳氢化合物装订技术,改进完整hNR基于LxxLL的亲和作用,但是并不改变装订的Src相互作用蛋白(Src-interacting proteins,SIPs)特异性结合框架。这种技术与传统的残基突变及化学修饰相比,既可以改变肽类配体的亲和性,又可以改变特异性。这就意味着碳氢化合物装订后,亲和性增加的SIPs可以特异性地与它们的亲本LxxLL基序竞争hNR阵列,因为它们具有和亲本完全相同的靶向特异性。
非典型孤儿受体DAX1和SHP组成hNR家族的NR0B亚家族。这两种受体缺少典型的DBD,并作为其他hNRs的辅阻遏物。DAX1和SHP结构域分别含有3 个和2 个保守的LxxLL基序,可以被hNR蛋白的AF-2结构域以激活构象的形式识别并结合。Broekema等[117]为探讨5 种DAX1/SHP LxxLL基序和人类基因组中发现的所有48 种hNR的AF-2结构域之间的系统性互作规律,分析这些基序靶向完整结构域排列的特异性和亲和性,设计出基于LxxLL的碳氢装订肽,可以利用该肽靶向特异性互作的每一个基序。基于结构域-基序复合物结构模型和结合力计算,获得每一个基序和完整hNR阵列的特异性互作,然后通过定量化测定这些基序之间结合力、相似性和相关性,构建了一个从基序到结构域的加权-靶向网络。
动力学模拟揭示基于LxxLL肽在非结合状态具有高度灵活性,因此,不利于被AF-2结构域识别与结合。碳氢化合物装订技术则可以用来帮助这些松散的肽活化螺旋构象,因此大大改善它们与hNR系列的亲和性结合。碳氢化合物桥设计用于标记活化的口袋结构域,并不破坏结构域和肽之间的直接互作。双重作用授予该装订对结构域和肽之间互作焓和脱溶仅具有中度影响,但可以分离和回收结合熵。对肽配体,其熵减可以大致看作一个常数,用以改良肽与完整结构域系列之间的绝对结合与亲和力,但是并不改变肽结合不同特异性结构域的相对亲和力。总之,装订肽可以被看作潜在的竞争对手,选择性靶向它们在DAX1和SHP蛋白中的亲本LxxLL基序,从而介导图5所示的特异性、复杂性互作网络,调节众多靶基因表达。

图5 基于结构域-基序复合物结构模型和结合力计算所构建的有向加权网络[117]Fig.5 Weighted source target network from 5 peptides to 48 domains[117]
3.6 AHR与核受体之间的复杂关系
AHR作为一种高度保守的胞内转录因子,可以被多种进入细胞内的外源和内生固有配体激活[153]。除可以进一步激活异源物质代谢酶之外,还参与造血系统、肝和免疫系统的发育与分化。越来越多的研究证明,它在免疫应答和诱发炎症并保持炎症状态等方面发挥重要作用。公认的观点是,AHR作为炎症分子的下游靶标,其转录受到炎症级联放大系统的控制。白细胞介素6(interleukin 6,IL-6)已被公认为早期炎症的标志,并直接影响AHR的表达。重要的是,该受体控制信号传导与转录激活因子3(signal transducer and activator of transcription 3,STAT3)激活,并和其他炎症介质互相配合。在选择性炎症环境下,一旦激活,AHR便与包括IL-6在内的靶标互作,发起一轮自发性炎症循环。这些证据表明,AHR是炎症的扩大和持续放大系统。就很多炎症介导者而论,它们以AHR作为至关重要的传感器和异源物代谢激活剂。AHR的敏感性环境配体可以解释某些芳香烃类化合物往往会引发某些个体的过敏和持续的炎症状态。因此,AHR可以被看作一个放大器,结合内源和外源配体与环境刺激并作出免疫应答。AHR的螺旋-环-螺旋基序是一个进化上非常保守的、遍在表达的生物传感受体。AHR具有多种配体,仅植物的吲哚和二苯乙烯苷以及动物源如前列腺素G2两类配体就有110 种以上。来自环境和化学合成的多环化合物有114 种,都可以激活AHR[154]。AHR连接内生和外源配体,调节一系列基因表达,包括相I细胞色素P450酶(cytochrome P450 proteins,CYP)系统(如CYP1A1、CYP1A2、CYP1B1)和相II酶系统(如谷胱甘肽-S-转移酶)[155]。AHR还和一系列相关细胞途径和基因互作,包括某些控制细胞周期的基因,如生长、分化、增殖和凋亡基因[156]。在无配体存在时,有热休克蛋白90、AHR互作蛋白、辅伴侣蛋白p23和一种蛋白酪氨酸激酶pp60src在胞浆中与AHR结合,使之保持静息状态。一旦有配体结合,该伴娘复合物便转位进入细胞核,发挥转录因子作用,亦即无活性的复合物裂解,AHR结合到AHR核转运蛋白(AHR nuclear translocator,ARNT)/缺氧诱导因子1β上,最终形成新AHR/ARNT复合物与靶基因的异源物质响应元件互作[157]。
败血症是由感染造成的系统性炎症综合征。败血症免疫应答的特点是同时激活促炎和抗炎两条途径。当败血症发生时,很多发炎细胞因子表达和激活是其显著特征。异源化合物受体作为化合物传感转录因子在药物代谢酶(drug-metabolizing enzymes,DMEs)的表达调控中发挥特殊作用。异源化合物受体介导败血症和药物代谢之间的功能调控。因为炎症细胞因子在败血症期间分泌并作用于异源化合物受体的表达和激活,从而影响DMEs的表达和激活。异源化合物受体也可以反过来作用于败血症的临床症状。LÜ Chuanzhu等[158]对这类受体和败血症之间的联系进行了详细综述。异源化合物受体(如孕烷X受体(pregnane X receptor,PXR)、AHR、糖皮质激素受体(glucocorticoid receptor,GR)和CAR)、药物代谢酶(如CYP1A、CYP2B6、CYP2C9和CYP3A4)以及药物运载体(如p-糖蛋白和多重耐药蛋白)都作用于败血症。AHR也是一种生理节奏性蛋白,也称为ARNT、专一蛋白结构域转录因子和典型异源化合物受体。虽然AHR并不属于核受体超家族,但是通过AHR传感化合物和调节DMEs的模式却非常相似于异源化合物核受体。除异源化合物传感功能之外,AHR在保持体内生理平衡,参与细胞增殖与分化、免疫应答、基因表达调控、激素代谢、炎症、免疫自识别及其对外部刺激的响应等方面发挥重要功能。AHR是炎症反应,特别是脂多糖诱导的炎症信号级联放大不可缺少的,与炎症因子的分泌密切相关[159-160]。值得注意的是,上调或激活AHR表达也可以调节抗炎症细胞因子IL-10的表达,下调促炎细胞因子IL-6和IL-8的表达,由此抑制炎症反应,保持免疫平衡,改善败血症的发生和预后。IL-10在败血症早期感染过程中通过多种免疫细胞抑制炎症反应,限制组织损伤和免疫病理作用[161]。IL-10受多种转录因子和信号通路的调节。不断积累的研究揭示,AHR是IL-10表达的关键调节者。Src酪氨酸激酶,作为9 种非受体酪氨酸激酶之一,是一种已知的细胞内靶蛋白,介导酪氨酸磷酸化。Src酪氨酸激酶可以诱导IL-10合成与分泌,下调促炎因子IL-1β、IL-6、IL-18和TNF-α的表达[162]。已知STAT3是脂多糖诱导炎症因子表达的信号传递和激活因子。STAT3通过结合到内含子4调节IL-10的表达,STAT3也是IL-10的下游调节因子。IL-10还可以以自分泌的形式激活STAT3[163]。LÜ Chuanzhu等[158]报道了Src酪氨酸激酶和STAT3两者磷酸化以及IL-10表达可以减少AHR敲除小鼠的炎症性巨噬细胞的衍生。这些结果都证明AHR对IL-10表达的非基因组学途径的正调控作用,而这一调控作用依赖于酪氨酸磷酸化介导的Src酪氨酸激酶和STAT3激活作用[164]。Zhu Junyu等[164]的研究证明,AHR对IL-10的正调控作用可以通过特异性Src酪氨酸激酶抑制剂降低,而且Src酪氨酸激酶抑制剂可以降低STAT3的磷酸化,说明Src酪氨酸激酶可以诱导IL-10上游的STAT3的激活。这些结果证明,AHR的激活可以增强Src酪氨酸激酶的磷酸化,然后,磷酸化的Src酪氨酸激酶(p-Src酪氨酸激酶)上调STAT3的磷酸化,磷酸化的STAT3(p-STAT3)接着正调控巨噬细胞合成与分泌IL-10。可见,AHR激活的炎症抑制作用可以改善机体对脂多糖的耐受性、调节免疫功能、降低败血症引起的死亡率。异源化合物引发炎症主要是通过炎症因子的释放,如IL-1β、IL-6和TNF-α,与此同时,异源化合物受体(如PXR、AHR和GR)接受相应配体刺激后也会靶向DMEs,从而启动代谢和排毒程序-药代动力学进程。越来越多的证据表明,异源化合物受体是药物代谢的重要介导者。显然,这完全证明了中国祖先“相生-相克”的哲学思想。DMEs主要用于药物代谢,将PXR和AHR用作炎症性疾病的治疗药物筛选靶点值得研究。
核受体可以针对环境变化快速激活/抑制免疫应答和适应环境变化的基因表达。然而,核受体对免疫系统的作用却缺少可靠的实验动物模型,以探讨核受体在体内和免疫系统的复杂互作。Diaz等[165]应用斑马鱼作为动物模型,考察了维甲酸受体(retinoic acid receptor,RAR)、肝X受体(liver X receptor,LXR)和负责胞浆传感AHR的免疫学作用。虽然同步激活作用并不能影响真实的靶基因表达,但是RAR诱导的il17a/f3可以由LXR和AHR发挥拮抗作用,反之,il22就会受到AHR,但不是LXR的拮抗作用。而且,维甲酸可以减少il10基因表达,这一表达进一步由于LXR激活作用而降低。因此可以用斑马鱼幼体进行组合性核受体激活作用研究。他们的研究还证明,LXR可以拮抗维甲酸诱导的细胞因子表达,从而为研究LXR和AHR调节免疫应答机制提供了一个实验动物操作平台[165]。
化学致癌物3-三甲胆蒽(3-methylcholanthrene,3MC)结合到AHR上,调控CYP1B1家族的CYP酶的表达,该酶参与环境污染所造成的致癌基因的激活作用以及雌激素的合成与代谢。3MC显示出诱导雌激素应答并结合到ERα上,刺激AHR和ERα互作功能。近来,已经报道GPER介导了某些内源性雌激素和环境化合物引起的雌激素样活性。Cirillo等[166]首先将3MC与GPER互作,确定其嵌入GPER,然后在SkBr3细胞和CAFs细胞建立了3MC激活EGFR/ERK/c-Fos信号,并通过AHR和GPER对两者的细胞信号传递的途径进行研究,结果发现,这些受体参与了CYP1B1和周期蛋白D1的上调表达,并在3MC的诱导下产生生长刺激作用。这说明3MC可能引发了AHR和GPER之间的生理和功能性互作,导致SkBr3乳腺癌细胞和CAFs细胞两者之间的互作和信号传递,从而诱发了GPER和AHR的细胞信号传导,推进了乳腺癌的进程。
4 GPER及其与配体互作
4.1 GPER的分布
最早关于克隆并表达了GPER的研究是在1996和1997年[32]。GPER的转录广泛分布于正常和恶性肿瘤患者的组织,高水平表达于心脏、肺、肝、肠道、卵巢和大脑[167]。一些原发性乳腺癌[168]和淋巴瘤[169]也有GPER表达。当然,这些早期的研究只限于对细胞系的研究。但是,近年来,GPER抗体的应用促进了GPER组织分布的详细调查。这些研究为我们提供了一个有关GPER多种功能的基本框架,其功能主要集中于雌激素依赖性生理响应。鼠科动物的免疫组化分析发现,不仅在各种组织中都发现GPER不同程度的表达,而且在雌、雄性动物中都有表达,只是表达量明显不同。总体上,GPER表达更多地集中在上皮细胞和肌肉相关性间叶细胞/成纤维细胞。Luttrell[170]通过免疫组化技术分析了成年大鼠,发现其GPER主要表达于下丘脑垂体-肾上腺轴、海马体和脑干自主核。这些研究结果表明,GPER对于生殖、分化、发育、肠道传感、吸收、代谢平衡、内分泌、神经、免疫,特别是各种上皮细胞的信号传递等具有重要作用。
4.2 GPER和核受体的结构与功能
雌激素和其他配体通过扩散进入细胞并结合到细胞核的ERs上,影响基因表达和细胞表现型。这种结合并激活相应受体的二聚体化进一步促进受体二聚体在DNA靶基因启动子区的直接互作,从而激活或抑制转录。ERs的两种亚型属于核激素受体超家族,当与各自的配体结合后具有转录因子功能[171]。核受体具有通常的结构和功能特点。典型的ER(现称之为ERα)含有595 个氨基酸残基和一个DBD、一个C-末端激素结合结构域(hormonebinding domain,HBD)。另外一种ER亚型,被鉴定为ERβ[172],这是因为它比ERα稍小一些,有530 个氨基酸残基[173]。而且两种ER在DBD结构域具有高度同源性(95%)。但是在A/B、D和E结构域很少保守。两种ER亚型的生理功能也有区别,特别是HBD具有相当少的同源性(53%),某些配体,如植物雌激素染料木黄酮就是一种ERβ选择性激活剂[174-175]。而且,ERβ缺少大部分C-末端F结构域。已知该区域是某些抗雌激素激活剂,如三苯氧胺的激活部位[176],由于这些A/B和F序列明显不同,其功能也有明显改变,所以ERβ高表达可能在三苯氧胺临床治疗乳腺癌所产生的抵抗效应上发挥作用。
ER介导的基因转录至少有两个不同的反式激活结构域,定位于受体氨基末端的A/B区(即AF-1)和羧基末端的E区(即AF-2)[177]。AF-1结构域不依赖于激素,而AF-2结构域则是激素依赖性结构域[178]。AF-1和AF-2两者都需要最大ER转录活性。但是,对于某些启动子,AF-1和AF-2则可以独立发挥功能[179]。有研究证明,ERβ的AF-1区激活作用和ERα相比几乎可以忽略不计,而AF-2的激活作用则具有可比性[180]。因此,ERα对ERE的激活作用可能超过ERβ,因为ERE控制的基因需要两个反式激活结构域的存在。抗雌激素,如三苯氧胺对这些基因只发挥单纯的拮抗剂功能,只需要ER的AF-2结构域介导的转录活性。相反,三苯氧胺则只能作为部分激活剂通过AF-1驱动转录,并不需要AF-2结构域[181]。激素结合和二聚化后,ERs以其DBD的高亲和性结合到DNA的特异性位点,也就是ERE上。当然,有些配体也可以通过ER结合其他靶基因应答元件的启动子区发挥功能。ERα和ERβ还可以形成异二聚体,从而改变对基因的反式激活作用。除这种经典的直接结合DNA的机制之外,这两种ER亚型还可以激活其他途径[182-183]。例如,AP-1应答元件间接受ER和AP-1转录因子c-Fos和c-Jun之间的互作调节[184]。这些转录因子所调节的基因参与很多细胞过程,包括细胞增殖、分化、运动和凋亡。因此,ER-AP-1互作可能具有重要的临床意义。研究显示,当ERα和ERβ表达时,三苯氧胺可以作为激活剂作用于AP-1应答元件所控制的基因。但是,与此形成鲜明的对比,E2在ERβ单独表达时,并不能激活这些基因[185]。这些发现说明选择性ERs调节剂往往具有不同的功能。
和其他生长因子信号途径之间的交谈是ER发挥细胞调控作用的另一个值得关注的途径。例如,酪氨酸激酶受体-EGF家族成员可以通过直接磷酸化关键性氨基酸残基激活ERs[186]。而且在ERs和胰岛素样生长因子(insulin-like growth factor,IGF)信号传递途径之间存在相当多的交互作用。ERs具有增加一系列IGF关键性信号分子表达的功能,而且IGFs反过来也可以激活ERs[187]。这种通过信号途径之间的交互作用-交谈可能对雌激素非依赖型发育和/或激素治疗临床抵抗具有重要意义。ERs的晶体结构揭示,在不同配体的刺激下,经历广泛和不同的构象改变[82,47]。ER配体结合结构域的变构作用取决于配体及其结合的本质属性。在雌激素-受体配体复合物中,ER配体结合结构域中螺旋12在配体结合时发生定位上的改变,在配体上形成一个保护性盖子。这种ER的构象改变暴露出一系列氨基酸以便与特异性辅激活因子结合。相反,当选择性ERs调节剂,如三苯氧胺或雷洛昔芬结合后则可以阻止螺旋12配体-结合口袋的形成。因此,了解螺旋12的精确定位对于ERs转录激活作用非常重要。
ERs并不能单独调控基因表达,很多组织特异性ERs可能依赖于其他因子影响转录活性。这些受体辅调节因子最早发现并得到研究的是核受体激活所涉及的调节因子,因为核受体激活常常受到其他受体和辅调控蛋白的干扰。这些辅调控蛋白不断被发现[188],其功能主要作为受体及通用转录机器之间的中间介导者,控制和调节ERs转录活性。辅激活因子或弱化(辅抑制)因子,是多种转录信号传递蛋白的辅助整合因子[189]。有些此类蛋白还具有内在的组蛋白乙酰化酶或脱乙酰化酶活性。一个过度简单的辅激活/辅阻遏作用模型被用来解释如何通过招募辅阻遏因子到受体复合物,从而使其从非活性状态转变为活性状态。一般而言,辅阻遏因子拥有组蛋白脱乙酰化活性,使核心组蛋白脱乙酰化,将靶基因的启动子紧密包装,从而关闭靶基因的转录。当配体结合以后,从转录复合物释放辅阻遏因子,使受体招募辅激活因子和具有组蛋白乙酰化活性的辅介导因子。这些乙酰基转移酶将乙酰基加到组蛋白上,从而失去与靶基因启动子结合的能力,开放靶基因启动子,适合于基本转录机器的结合、识别与转录。然而,似乎还有更多的酶活性与很多受体辅调控蛋白活性有关,包括蛋白酶、泛素连接酶、ATPase和激酶等活性[190]。
在经典的ERα信号途径,E2结合并诱导ERα二聚体化,然后结合ERE[191],调节基因表达[192]。配体结合ERα后,招募辅调节因子复合物和不同种类的酶一起修饰和重构核小体,调节基础转录装置[193]。ERα含有核受体超家族的结构域AF模块,包括位于N-末端结构域的AF-1、一个DBD和一个C-末端LBD。在LBD还含有一个AF-2表面。重要的是,核受体作为一个支架蛋白和配体一起通过LBD微妙的结构变化控制受体-蛋白和受体-DNA互作。这些微妙的结构变化如何引起细胞信号传递和活化功能的细微变化,这一问题到目前为止仍然是一个富于挑战性的研究课题,因为其结构的细微变化尺度可能小于1 Å。换言之,配体与受体的互作所引起的变化可能在单一原子的变化就足以产生不同的信号输出,该变化尺度和晶体结构的噪音处于同一个水平。所以Nwachukwu等[191]引入了一种系统生物学和晶体结构相结合的方法解决这一关键性难题。他们测定了联系紧密的相关配体的结构与功能、它们与特异性分子骨架之间的关系及其在LBD组装晶体结构中的变化。这些方法相结合,能够鉴定配体-特异性结构扰动所造成的不同信号输出;但缺点是,只能定性分析,而且依赖于结构模式的视觉识别;因此又引入了一个定量化、无偏差方法,这种方法能够对配体与不同的骨架,而不是紧密相关的骨架互作所产生的联动变构作用进行分析。此外,他们还通过ERα的晶体结构与变构信号传递之间的关系,解决了ERα-LBD复合物与环境化合物(拟雌内酯、农药双对氯苯基三氯乙烷和玉米烯酮毒素)所引起的配体对LBD结构的微妙扰动;更有趣的是,对42 种人工合成配体对ERα的LBD扰动所介导的转录活性、细胞增殖作用的统计分析结果显示,它们之间有很好的相关性,并且通过分析这种相关性可以进行结果预测。这些作用主要发生在原子间的扰动,处于亚Å(the sub-0.5 Å)尺度和X射线衍射测定化学结构的噪音范围,但是却决定了细胞信号的特异性传递和应答。推测这些原子间互作之所以能够决定特异性信号传递、输出和响应,主要是由EE/ERα的LBD结构域的原子间扰动所产生的联动变构,以及之后的基因组学辅助调节因子所决定的。
4.3 GPER结构及其与配体之间的互作
GPER与不同配体之间的互作在药物筛选、雌激素类化合物结构和功能评价等方面具有重要理论意义和应用价值[194]。有趣的是,这些化合物,如G1和G15具有明显相似的化学结构[195];G1拥有一个附加的乙酰基,负责诱导GPER构象改变,并通过其氧原子空间和/或极性效应产生激活作用。极性效应存在于所有GPER的激活剂配体(E2、G1、三苯氧胺、氟维司群等)中,但是所有拮抗性配体(G15和G36)都没有。G36以甲基取代了G1同一位置上的乙酰基[196],E2和氟维司群则拥有一个相似的核心化学结构[197],但是氟维司群还具有一个附加的疏水长链。在E2和三苯氧胺之间除上述提及的氧原子之外,没有化学相似性[198]。尽管如此,两者都能够与GPER互作[199],这无疑强化了GPER可以接受多种化合物刺激的机制有别于ERα/β。因此,深入了解GPER的结构和结合位点将有助于新型药物的设计、筛选与评价[200]。
最近的分子动力学模拟和镶嵌研究使GPER的结构元件及其作用机制得到进一步揭示,蛋白结合位点的结构变化及分子运动[201]可以为生殖、神经、内分泌、免疫、心脑血管系统相关疾病治疗提供一个新药设计平台[202]。Méndez-Luna等[194]提供了一个GPER的三维结构模型,然后通过分子对接法探讨GPER结合口袋及其与配体的识别特性,调查关于激活和钝化作用机制。为了实现对有关结构的了解,应用一些已知的激活剂(E2、G1、三苯氧胺和氟维司群)和拮抗剂(G15和G36)进行研究。由于这些蛋白隐藏在细胞质膜内,用传统的实验方法难以得到其三维结构。一般X射线衍射或核磁共振波谱学研究需要利用其水溶性获得单晶[203],而这一般只适用于水溶性蛋白[204]。有些GPCRs可以用激活剂或拮抗剂、抗体、突变等进行处理,以提高其结构稳定性[205]。因此,Contreras-Romo等[206]参考现有的GPCR实验数据和已经建立的其他模型,构建7TM 3D计算机模拟模型,通过分子对接计算机模拟并结合已有相关结构信息,对有关配体与GPER识别过程进行研究,结果表明,所有配体和GPER互作都有不同的自由能变化,分子对接和分子动力学模拟结果显示,E2、三苯氧胺和氟维司群与受体互作的结果随时间趋向于负值。G1-GPER复合物是其中最稳定的,通过受体-配体识别的结构数据表明,G1和G15之间均方根误差最小,这是由于G1和G15化学结构的相似性,补偿时间为14 ns,而其他复合物的补偿时间为70 ns。显然,只有G1和G15保持相同的结合基序,G1和G15相比只有一个额外的乙酰基,因此这个额外的基团就是负责诱导受体适应配体活性的构象。除G1、G15和G36以外,其他配体都具有另外的残基赋予它们更多化学基团,说明该蛋白具有一个可以适应不同配体以及潜在的相邻结合位点[207]。应用分子对接研究还发现,配体-蛋白形状可以通过变化来加以适应[208]。在这些结合位点中,Phe278和Phe208是已建立了配体与芳香环部分互作的氨基酸残基[209],而一系列杂环氢原子也显示出形成氢键的特性,说明在与大分子互作时,可以与更多的功能基团形成氢键。芳香族氨基酸簇(F206、F208、F223和F278)揭示了化合物GPER-L1和GPER-L2的生物活性是由于它们作为激活配体,可以与GPER的不同构象和不同氨基酸残基互作[209]。Cys207与G306互作,可能是涉及开关或活性状态切换的关键性残基。识别G1和G15的氨基酸残基还可能参与结合位点的阻断,或许是Cys207残基诱导GPER构象的改变后,通过G1的乙酰基产生阻断作用。无论哪种情况,上述假说的确得到了该研究结果的证实,而Cys207和E275也参与了G1和GPER的互作,其他配体激活剂的测定结果亦如此。总之,这些研究有助于我们对GPER配体结合口袋的调查和了解。应用已知的激活剂和拮抗剂可以推测,GPER含有一个较大的配体结合口袋,并通过构象改变适应各种不同配体的结合,但是决定性状态开关或切换位点尚待深入研究。
Cao Linying等[210]也用分子对接方法模拟了30 种多溴联苯醚(poly brominated diphenyl ethers,PBDEs)和OHPBDEs与GPER的互作。他们成功建立了受体的同源结构模型,模型中配体结合口袋被定位于跨膜螺旋2、3、5、6和跨膜螺旋TM7深缝隙之中。首先,3 种已知对GPER具有亲和性的配体化合物(E2、G1和G15)以及一种无活性的化合物(α-E2)嵌入GPER中用来验证该模型并作为感兴趣PBDEs/OH-PBD的对照。结果表明,E2通过其17-β羟基嵌入GPER延伸到跨膜螺旋6,与Asn276形成一个氢键;与E2相似,G1也通过与Asn276互作,通过乙酰基的氧原子形成一个氢键;G15和α-E2则不能和任何氨基酸残基形成氢键。然后,分别将PBDEs和OH-PBDEs嵌入GPER配体结合的口袋,结果发现,在这些化合物中,有9 种OH-PBDEs与GPER通过其羟基形成氢键。但是,不同于E2与Asn276形成氢键,OH-PBDEs是通过Ser134、Gln138、Phe208、Glu218、Glu275、His282依赖于OHPBDE和His307形成氢键。虽然OH-PBDEs可能通过多种机制发挥其雌激素作用,但是以上研究结果明确显示,只通过其中的一种直接结合机制发挥胞内信号途径的激活作用。该研究结果强调,OH-PBDE主要是通过干扰雌激素传递信号的发挥功能。
4.4 受体互作网络与表观遗传修饰
生物通过营养吸收及合成与分解代谢为其发育、分化和各种复杂生理活动提供“建筑材料”和能量。显然,所有的生命活动都离不开这些最基本的“新陈代谢”活性。正因为如此,几乎所有以往的教科书都把这些不可或缺的代谢和生理活动控制基因称为持家基因,否则称为奢侈基因。因为持家基因大多数编码了基础代谢或中心代谢途径中的酶,所以这些酶往往属于组成型(一直表达的)酶,只适应于某些条件才表达的酶则属于诱导酶。然而,这些称之为组成型酶的基因是否在基因表达水平上不受调控呢?显然不是。生物对食物和营养的需求受到环境、饮食和营养的限制和调节。Emmett等[211]综述认为,基因表达,特别是持家基因表达的精确调节对于哺乳动物生长、发育、生理、内平衡及其对环境的适应极其重要。越来越多的研究结果证明,基因表达调控首先通过序列特异性转录因子进行,这些序列主要定位于顺式增强元件[212-214]。而这些转录因子则往往依赖于配体的结合,例如核受体[215-217]。当核受体与配体结合后,还需要招募不同的转录辅因子,组装成一个复杂的转录复合物,从而调节基因的转录和表达。在某种程度上,转录辅因子的招募进一步调节染色质的结构,包括通过HATs对组蛋白乙酰化和HDACs对组蛋白脱乙酰化[218]进行写入或擦除[219-220]。HAT活性往往是转录辅因子的固有活性,可以通过将乙酰辅酶A的活性乙酰基转移到组蛋白氨基端赖氨酸残基的ε-氨基上,促进基因表达[221-223]。HDACs和HATs的功能恰好相反,它通过去除组蛋白尾巴上的乙酰基,抑制基因表达。大量研究证明具有相反酶功能的精确控制构成发育和生理活动,是合成与分解代谢、物质和能量代谢相关基因表达的关键。哺乳动物基因组编码了11 个HDAC亚型,从而构成了HDAC超家族,这些酶发挥特异性和交叉功能两种作用,分布在细胞核与胞浆中。第1个HDAC分离鉴定并命名为HDAC1[224],其编码基因在哺乳动物中很保守,共发现11 个成员,构成了HDAC家族[225]。这些蛋白的编码基因和HDAC1具有高度同源性,并分类为:第I类HDACs,包括HDAC1、HDAC2、HDAC3和HDAC8;第II类HDACs,包括HDAC4、HDAC5、HDAC6、HDAC7、HDAC9和HDAC10。所有第I和II类HDACs都含有一个保守的脱乙酰化酶结构域,而且酶催化功能都需要锌的参与,虽然第II类HDACs的HDAC4、HDAC5、HDAC7和HDAC9的酶活性尚难证明[226]。第IV类只有一种,即HDACII,在脱乙酰化酶结构域也存在一个与第I和II类同源的保守序列。但是在结构上有区别,类似于尼克酰胺腺嘌呤核苷二磷酸依赖型的第III类HDACs(即已知的Sirtuins),但是没有第I和II类HDACs所具有的一般结构[227]。显然这些依赖于核小体的多种修饰系统构成了人类饮食、生活方式,不断在染色体上写入、读取、修改或删除,并通过表观遗传的形式形成记忆和遗传进化的分子机制。
5 复杂多样的GPER和ERs受体传感系统
5.1 GPER的毒性和有益双重作用
GPER接受胞外配体的控制,通过其七跨膜结构控制细胞内基因组学和非基因组学信号途径,其中非基因组学途径主要用来控制离子通道,从而快速控制神经、内分泌应答;基因组学途径则通过核受体网络,特别是ERα/β将信号传递到细胞核内控制众多基因的转录、剪切、翻译、加工和转运。Yang Jiali等[228]综述了由ERs所引发的生物学功能。
5.1.1 ERs的多种生理功能
ERα对于生理发育和正常代谢具有特殊意义。敲除ERα将导致不孕、泌乳激素降低、乳腺不成熟、骨骼减小、胰岛素抵抗和肥胖。然而,敲除ERβ则可能导致生育能力下降、卵母细胞减少、不再肥胖,并伴有血清胆固醇、瘦素和血糖浓度增加[229]。在骨骼和代谢方面则无宏观异常表现[230]。ERβ作为雌激素信号途径的显性负调节因子,显示出对ERα介导的转录激活作用的抑制效应,因为雌激素正常的信号途径是通过ERα信号途径。高水平的ERβ表达在临床治疗中主要是作为一个良好的预后标志,可见ERβ可以抑制过度的雌激素作用。ERs在免疫调节中也发挥重要作用,既可调节先天免疫,亦可调节获得性免疫[231]。在E2低于生理水平时,ERs,特别是ERα一般都会促进I型干扰素分泌和促炎细胞因子表达。而在E2水平过高时,就会产生抗炎症应答,ERβ信号传递可能偏向于负责对促炎细胞因子的衰减作用[232]。ERs的另一个重要作用是神经保护。ERβ在人类主要分布于海马体[233]。在更年期,内生合成的E2在海马体中足以结合并激活ERα。当E2处于高水平时,ERβ则展示出正调控活性以改善记忆。ERα/ERβ的比值对于调节行为、成熟和衰老非常重要。ERβ表达于中缝背核的羟色胺能神经,调节色氨酸羟化酶,参与血清素(5-羟色胺)的合成。选择性ERβ激活剂可以恢复更年期所造成的色氨酸羟化酶的丢失,减轻抑郁,防止情绪失控[234]。
5.1.2 植物雌激素的有益和有害作用
植物雌激素类化合物正是由于具有相似于雌激素配体的结构,发挥对ERs的调节作用。植物雌激素类化合物的安全性已存在较长时间的广泛争论,大多数文献认为植物雌激素有益健康,而且没有副作用。然而,也有一些有关植物雌激素有害作用的报道,例如,有研究表明富含植物雌激素的大豆配方会显著增加患子宫肌瘤的风险[235]。在一个对更年期综合征临床治疗的荟萃分析中发现,植物雌激素中度提高了胃肠副作用[236]。过高剂量的植物雌激素可能会阻碍乳腺发育,而低剂量又会产生相反的作用[237]。年龄、健康状态、肠道微环境和剂量是影响植物雌激素正、负作用的主要因素。
早在2007年,Wuttke等[238]就依据当时的大量数据,对植物雌激素,特别是异黄酮类植物化学物的正、反两方面功能进行分析和综述。首先,它们的抗动脉粥样硬化作用具有很大争议,因为尚无精确的研究发现其对体脂有任何有益作用。更重要的是,关于来自大豆或红三叶草的异黄酮对乳腺或子宫内膜的有益或有害作用也存在争论。事实上,只有在童年和青春期持续食用异黄酮类食品的人群才表现出大豆制品的防癌、抗癌和预防乳腺疾病有益作用,这一点已经得到了科学证实。或许这也是所谓“日本人现象”的真正原因[239]。因为日本妇女患乳腺癌的比例低于西方人,甚至低于西方出生的日本人,主要原因可能是在童年和青春期持续食用含高异黄酮的豆制品所致。当毫无饮食异黄酮经历的更年期妇女食用异黄酮时,这些植物雌激素显然具有服用雌激素类似的作用和副作用,如刺激子宫内膜和乳腺组织增生,从而对这些器官或组织带来病变风险。因此当对食品的功能性成分,特别是植物雌激素进行功能评价时,应该考虑其生活和饮食经历。
蛋白质需求在日本主要是由大豆制品来满足,但是在西方国家大多数是靠肉、蛋、奶。因此,科学家在寻找功能性成分时发现,大豆在中国、日本和东南亚对乳腺具有潜在性保护作用。很快,许多研究推测,异黄酮、染料木黄酮和黄豆苷元作为黄豆等食品中对乳腺具有保护性作用的有效成分,这些植物雌激素可能就是导致“日本人现象”产生的功能性成分[240-241]。这一推测夸大了豆制品和异黄酮类的治疗和保健功能,目前并没有确切的疗效和临床证据。由于雌激素缺乏存在骨质疏松或动脉硬化的风险,绝经后妇女被推荐增加异黄酮类植物雌激素服用或添加了异黄酮的功能性食品。然而,来自大豆或红三叶草的异黄酮是否具有治疗作用、有无毒副作用、是否与经典雌激素同样具有双重作用,Wuttke等[238]对此进行了详尽的整理和总结分析,剔除了远高于食用此类产品后血清中所能达到异黄酮浓度进行统计分析,结果表明有益作用并不明显。对于一些具有统计意义的报道进行分析,虽然也证明植物雌激素类化合物存在一定的有益作用,但是其诱导乳腺、子宫内膜增生,甚至患乳腺癌的风险显著增加。换言之,植物雌激素类化合物发挥作用并不明显,即使能够发挥作用,也与经典的雌激素相似——具有双重作用。图6是从结构上对比E2和染料木黄酮的相似性,揭示出两者具有相似的OH基团、距离和定位,可见二者完全可能与ERs或GPERs的配体结构域进行识别与互作[238],说明其发挥生物学作用的途径也是通过体内雌激素E2的受体信号途径,同样具有有益和有害双重作用。

图6 E2-17β和染料木黄酮的结构相似性Fig.6 Structural similarity of estradiol-17β and genistein
5.2 ERs的双向调节作用
所谓“双向作用”或“双向调节作用”存在多种含义和不同解释。1)不同剂量发挥完全相反的调节作用。例如,Xiong Xingui等[241]研究表明,传统中药枳壳厚朴汤具有诱导胃动力的双向调节作用。枳壳和厚朴同时使用可以在某一剂量协同提高胃动力,低剂量时表现为增加胃动力,高于某一剂量时则转而抑制胃动力。枳壳厚朴汤改善胃动力主要是作用于毒蕈碱受体,其次作用于α受体。Xu Zhixiang等[242]利用MCF-7细胞系研究槲皮苷的细胞增殖、迁移、浸润、细胞周期、凋亡和氧化应激作用,结果显示,槲皮苷可以对细胞的行为产生剂量效应,依赖于活性氧-调节p53信号途径。槲皮苷在低浓度时促进细胞增殖并抑制细胞凋亡,而在高浓度时则相反,诱导细胞凋亡。此外,槲皮苷在低浓度可以显著抑制抗雌激素三苯氧胺诱导的MCF-7抗增殖作用,而高浓度时则可以协同促进细胞凋亡。实时定量聚合酶链式反应分析结果表明,槲皮苷在三苯氧胺诱导的抗细胞增殖作用中,通过调节涉及细胞转移、细胞周期和凋亡的ER途径双向调节靶基因的表达。2)在相反的两个方面发挥调节作用。Liu Haixin等[243]综述了雌激素对激活固有新血管生成的调节作用,但是由于其潜在的致癌作用不能用于治疗。植物来源的雌激素则可能提供一个替代性选项,但是其对新血管生成效应尚缺少深入分析,甚至互相矛盾。基于现有文献进行总结分析得出如下结论:促血管生成植物雌激素主要功能是对心血管的保护作用,而抗血管生成植物雌激素则主要在癌症预防和治疗中发挥作用。这种双向调节作用展示出靶标选择性和ERs依赖性。可通过血管生成相关基因转录检测植物雌激素对ERα和ERβ的反式激活特性。因为当不同的植物雌激素作用于ERα和ERβ时,对血管生成显示出相反的信号途径。作用于ERα的植物雌激素激活或抑制一些血管生成相关基因,使血管生成作用活化升高,而作用于ERβ的植物雌激素调节血管生成相关基因转录的结果则导致血管生成抑制。因此,植物雌激素对ERα和ERβ的选择性作用或许可以成为调节促进或抑制血管生成过程之间平衡的关键。有趣的是,白藜芦醇显示出既可以抑制也可以促进血管生成的双重作用[244]。3)还有一种双向调节的现象:当自身雌激素分泌过低时,可以通过服用适当的植物雌激素上调;而自身雌激素分泌过高时,服用同样的植物雌激素对其下调。图7显示了这种调节可能的体内作用机制。假设固有的雌激素E2对受体作用可以获得百分百的信号输出,则植物雌激素对这些受体的作用为固有雌激素信号输出的60%。在自身雌激素分泌过多时,通过服用植物雌激素,如异黄酮,可以和自身雌激素形成竞争ERs的关系,使得这些接受植物雌激素刺激的受体所输出的信号减少40%,总输出小于100%;但是当固有雌激素低于正常生理水平时,植物雌激素又可以补足到60%,整体上表现出双向调节的效果。

图7 植物雌激素对ERs的双向调节作用Fig.7 Bidirectional regulation of phytoestrogens on estrogen receptors
5.3 华夏民族的饮食智慧
中国具有数千年的农耕饮食文化和中医药理论、经验,按照《灵枢·营卫生会》中的理论,用现代生命科学概念进行总结,植物性食品和中草药对机体的免疫作用主要表现为:“阴”即上调免疫”、“阳”即下调免疫、“热、温”即促进机体分解代谢、“平、凉、寒”即降低分解代谢,并根据经验将其与“辛、甜、酸、咸、苦”五味相对应,从而创造了中医、药、食品的生理、免疫、代谢通过“相生-相克”调节,达到阴阳和谐、生理和代谢平衡以及营养均衡的理论与规则[245]。中国的平衡膳食理论认为,长期食用“偏性”食品就会造成机体免疫和生理代谢失衡,从而导致疾病;中医药和保健食品的作用就是以偏纠偏,通过“寒者热之”或“热者寒之”,维持饮食平衡。在植物雌激素类化合物和植物化学物的研究中,存在对受试群体饮食和身体背景复杂性估计不足的一个关键性问题,因为饮食营养受环境、气候、生活条件、生活方式、饮食文化、营养状态和遗传背景等诸多复杂因素的影响。表观遗传学证据表明,几乎所有细胞的生存和营养环境都可以通过DNA和组蛋白的多种修饰方式将这些营养代谢过程“写入”染色质中,并可以根据环境变化,特别是营养状态、生理和代谢需求情况不断地对染色质进行“写入”、“阅读”或“擦除”。可见,如果按照西方的“逻辑实证主义”哲学方法研究植物雌激素的健康作用时,所得出的结果必然会因为其遗传学,特别是饮食和生活方式所积累的表观遗传修饰的不同而不同,甚至完全相反。正如上述“日本人现象”所示。事实上,日本乃至东南亚,与中国不仅有非常相似的饮食文化、饮食结构和生活方式,在中医药和饮食与健康哲学上也有千丝万缕的联系。“阴、阳”和“热、温、平、凉、寒”的“平衡与纠偏”思想已经深入到每个人的生活习惯和方方面面。这就是以中医、药和饮食“平衡与纠偏”理论为基础的饮食智慧,或许这才是个性化营养的必由之路。
6 GPER研究需要进一步解决的关键科学问题
尽管雌、雄激素受体可以直接通过核受体网络将信息传递到细胞核内,调节染色质的修饰、转录以及剪切、翻译等基因表达调控的基因组学途径,但是其非基因组学途径则必需依赖于GPER的离子通道“开/关”控制机制。另一方面,虽然激素受体广泛分布于几乎所有组织、器官和细胞以及细胞膜、内质网膜、核膜等膜系统,从而将细胞内外的内源性激素信号传感并传递到细胞内,但是对于外源性激素信号的传感和传递则需要依赖于GPER。近些年来,虽然GPER得到了广泛和深入的研究,但是仍有一些期待解决的关键科学问题。
6.1 GPERs的精细结构有待解析
GPERs作为一种GPCRs超家族成员,它和配体的互作动力学,特别是精细结构尚未解析;结构与功能的关系尚需深入研究。作为膜蛋白,要得到单晶并进行X射线衍射分析并不容易,冷冻沉淀电镜技术的兴起不仅为这些膜受体提供了解析其精确结构的新途径,而且可以通过与配体共沉淀,研究和推测其结构与功能的关系。但是和酶与底物所形成复合物的情形相似,受体和配体互作、识别、变构以及向细胞内传递信号的过程往往发生在更短暂的瞬间。因为酶和底物结合需要把底物变成产物,而受体与配体互作只需要瞬间的变构并激活胞内信号放大作用即可。将这个过程描述为“结合(binding)”并不恰当,更应该精确地描述为“敲击键盘(tapping the keyboard)”:相当于环境信号通过“敲击”细胞膜受体“键盘”向细胞内输入信号的过程。笔者认为用“敲击”来取代结合非常重要,因为这两种描述至少有两个本质区别:1)“敲击”更短暂,而且可以反复进行多次,而结合则需要“结合力”和 “过程”,对酶来说,需要把底物变成产物这一过程;而且底物一旦变成产物就不会再结合;2)配体通过受体进行跨膜信号传递的过程更相似于酶的活性调节,而不是酶的催化过程,前者只需要使受体变构的作用,而后者则需要结合-催化使底物变成产物-释放产物的过程。之所以要区分这一概念,是因为到目前为止,能够得到精确解析的受体,特别是能够对受体-配体互作进行精确解析的几乎全部局限于配体和受体可以紧密结合,并可以一起冷冻沉淀的情况。然而,大多数受体-配体之间的互作并不是通过紧密(或稳定)结合,而是通过一个微小的和瞬间的结构变化,ERs和GPER便是如此。并且,从传递信号效率方面判断,传递信号效率(或灵敏度)越高,受体-配体互作的时间越短,同一配体“敲击”同一受体的次数也越多。显然,受体,特别是GPER的这些特性构成了解析配体-受体精细结构与功能关系的最大挑战。
6.2 GPER与配体之间的互作参数与动力学问题
事实上,配体-受体识别和变构,并将此变构作用联动到细胞内和G蛋白相偶联,通过GTP高能键释放能量驱动信号级联放大是一系列动力学变构过程。所以,要真正深入探明其细胞的分子生物学机制,单从受体结构,体外操作是不可能实现的,必需在研究过程中引入受体-配体互作、受体胞外结构域与配体互作、胞外结构域与胞内结构域之间联动变构、胞内结构域与G蛋白偶联以及激活胞内信号放大和传递的全过程。从传感器的角度来考虑,这不仅需要通过构建受体传感器来检测受体-配体互作动力学,还需要构建细胞或组织传感器来研究胞外结构域与配体互作通过联动变构激活胞内信号的放大与传递,并进一步通过基因组学途径传递到细胞核内从而控制基因表达的过程;以及如何激活非基因组学途径,控制细胞相应的离子通道开关,在细胞-组织-器官之间传递电化学信号,或通过电信号在神经网络中发挥作用。可惜迄今为止尚未见有关GPERs传感器及其与配体互作动力学的研究报道。
6.3 GPER用于食品及其化学成分的功能性评价
在食品及其化学成分的功能性评价方面一直充满争议。争议的焦点主要集中在:营养是食品的本质功能,所以如果从营养的角度评价食品,只需要考察能量、蛋白和各种维生素是否可以满足不同人群的营养需求。但是当满足人类不同群体的营养需求,特别是各种营养配方都已推出以后,人类由于营养过剩所造成的疾病并未得到有效控制,反而与日俱增。不得不直面一个不争的事实是:食品除可以为机体提供营养功能以外,还有其他功能。但问题是:其他功能是什么?如何对食品营养以外的功能进行评价?大部分研究参照医、药领域的实验动物模型和细胞系进行评价,但是,药物和食品的最大区别是药物是用于治疗疾病,而食品则是提供营养以保证机体健康和生存。所有的药物都有不同程度的毒性,并且到目前为止,尚未发现任何一种动物的生活习惯、饮食结构、遗传背景和人类相同,甚至相近。
6.3.1 食品营养学的出路
随着植物化学物和植物雌激素对人体代谢、生理、老化、免疫和内分泌调节作用的不断揭示,食品不再仅仅被作为营养为人类生存提供支撑,营养配方也不再能够为现代人的健康提供可以信赖的保障。随肥胖病、糖尿病和心脑血管疾病的蔓延,不难发现,同样的饮食对不同饮食背景、生存环境、生活方式、遗传背景的人群产生的后果不同,而且这些差别存在于人类生活的方方面面,随时可见。所以有科学家提出了营养基因组学的概念。基因组学研究表明,虽然不同民族人群几乎在基因编码上不存在任何差别,但是人类个体之间却存在巨大的单核苷酸多态性(single nucleotide polymorphism,SNP)。人类个体之间在营养吸收、需求,特别是植物化学物或植物雌激素调节方面的差别可能正是由基因组学水平上的这些SNP造成的。然而,很多科学家开展对营养基因组学的集中研究时,表观遗传学却很快以大量的研究结果证明,人类的饮食结构、生活方式,特别是对营养的摄取、吸收、合成与分解代谢,以及解毒、排毒过程可以通过甲基化修饰DNA和组蛋白,甲基化、乙酰化、磷酸化、琥珀酰化、棕榈酰化、巴豆酰化修饰核小体组蛋白“写入”、“阅读”或“擦除”这些信息,这些对生活方式和饮食习惯的染色质“记录”,不仅能够形成长期的记忆和积累,而且在生存竞争和进化中具有重要的选择优势,构成了表观遗传的主体。而这种染色质修饰和“记录”系统正是通过GPERs和包括ERs在内的核受体网络得以实现的。在这个意义上,食品营养及其功能评价的根本出路可能并非营养基因组学,而是GPERs和核受体所形成的网络。
6.3.2 “中华饮食理论”对食品功能评价的贡献
中国人评价食品功能的方法是在长期饮食经验的基础上归纳总结食品和中药的基本规律。所有的食品按照其免疫学和生理、生化作用,将增强免疫(促进免疫应答),具有“滋阴”作用的食品确定为“阴”性食品;将下调免疫(抗炎性),或具有壮阳作用的食品确定为“阳”性食品;在此基础上,进一步按照食用后对机体代谢所产生的作用分为“热、温、平、凉、寒”5 种属性,促进分解代谢的为“热”或“温”,促进合成代谢的为“寒”或“凉”,不影响代谢平衡的为“平”性。但是,由于这5 种属性难以定量化,而且并不严格、分明,所以纳入了很容易品尝的“苦、咸、酸、甜、辛”五味[14]。
6.3.3 “阴、阳”、“热、温、平、凉、寒”属性、“五味”及其受体
中国人所谓饮食中的“阳”主体上对应雄激素受体,“阴”则对应于ERs。值得注意的是,人类对周围的物质世界主要是通过嗅/味觉受体传感和认知;食入胃肠道后则进一步通过胃肠黏膜系统的多种受体进行传感、识别、免疫调节与记忆。肠道中的味觉受体,如T1R1(氮营养传感受体)、T1R2(碳营养传感受体)、GPR120(游离脂肪酸传感受体)等,近年来已经得到大量研究,其他受体如免疫相关受体、CD45、细胞因子和趋化因子受体也积累了不少研究[246-247]。特别是ERs,如ERα/β、GPER等均分布于肠黏膜系统,发挥免疫、代谢、内分泌调节功能[248],并构成肠-脑[249-250]、肠-肝[251]、肠-肺[252]、肠-肾[253]、肠-骨轴[254]等。
6.3.4 GPER与食品功能评价
显然,食品除为机体提供各种营养外,还具有多种功能。其中最重要的是为满足机体营养需求所形成的营养摄入、吸收和控制系统的形成和进化。目前的研究结果表明,该控制系统主要是通过食欲调节。饥饿时,食欲增强,食之有味,鼓励进食;当饮食超过胃肠道消化和吸收能力时,食欲下降,食之无味,停止进食。这是生物摄取营养的基本法则。所以饮食的基本功能是营养摄取及其控制。但是,对于动物源食品,因其主要成分相对简单,主要为蛋白质、核酸、淀粉(肌肉)和脂肪酸类宏营养,其他如维生素、矿物质和微量元素只能满足人体主要营养需求。但是植物源食品因其化学成分的巨大多样性,除可以为机体提供基本营养需求外,还可能发挥多种复杂的生物功能。这不仅构成中医药治病救人的基础、现代医药靶向化合物的源泉,同时也成为饮食健康、“不治已病治未病”的基础。然而,面对多达数万种植物化学物,每一种化合物都具有多种功能,多种化合物之间互相叠加、协同和拮抗作用;此外,剂量效应、人类个体之间的种属、遗传、环境、饮食结构和文化上的差异,会进一步增加其多样性和复杂性。彻底阐明这些化合物的功能几乎是不可能的。然而,基于中医的传统理念提供了一种可行的思路:系统评价这些化合物的混合物,也就是“全食品”对人类营养与健康的功能。作为GPCRs受体超家族成员,GPERs通过传感胞外雌激素或植物雌激素类化合物向胞内传递两种类型的信号:1)通过非基因组学途径控制各种离子通道开关,用来快速传递神经、代谢、生理和内分泌信号,控制食欲、进食行为和生理代谢平衡。2)通过ERα/β等核因子互作网络向细胞核内传递信号,通过NCoR和SMRT复合物“写入”或“擦除”核小体修饰,启动或关闭相关基因(包括细胞因子、趋化因子和激素)转录,由这些机体内的通讯分子调节和控制整个机体的细胞通讯网络和代谢网络。GPERs和多种核受体已作为药物筛选靶标,并取得很多研究成果,但是在食品功能评价方面则尚未得到应有的重视。这是因为:1)药物筛选早期并不需要对每种配体进行定量化评价,而定量化方法目前只能通过细胞系及其信号途径,多数只能通过基因组学途径中分子标记才能实现;2)GPERs虽然在医药领域得到了广泛了解,但是食品科学界则并未熟知;3)在技术层面上,有关受体传感器的研究成果尚十分有限,即使有些成功的报道往往缺少后续研究,或未能实现可以接受的选择性和可重复性,难以推广和应用。笔者所在的实验室近年来在受体传感器方面进行了较为系统的研究[255-257],在受体基因合成、表达、分离、纯化、纳米受体传感器、细胞和组织传感器、受体在不同种属之间的自组装、受体传感定量化和动力学研究等方面取得了系列进展,这些技术为人类GPERs受体表达、传感器研制及其对不同雌激素和植物雌激素类化合物的定量化测定与功能评价奠定了基础。
7 结 语
越来越多的研究结果表明,植物化学物、植物雌激素和中草药有效成分具有大量交叉和一致性。这些结果一再证明了植物化学物不仅是药物筛选、中药配伍、中草药有效化学成分鉴定及其定性、定量研究的基础,同时也为食品功能性化学成分的定性和定量评价奠定了基础。另一方面,对植物化学物的功能评价及健康作用研究所得出的结果差异很大,同一种植物化学物对不同的调查群体、不同的剂量等调查结果往往充满变数,甚至恰恰相反。这些研究结果的差异性和复杂性说明,不同植物化学物的剂量、配伍、食用途径可能发挥不同的作用;更重要的是,同一种植物化学物可能由于食用者的遗传背景、饮食习惯、生存环境、生活状态、年龄结构和性别所形成的表观遗传基础不同而不同。显然,要真正评价这些食品或植物化学物的功能,揭示其规律,需要一个可以定量化、标准化的检测平台,而GPERs正好可以提供这样一个平台,通过人类GPERs受体传感器的构建、自组装到各种细胞、组织或器官,研究不同植物化学物或植物雌激素类化合物的动力学规律和重要参数定将在食品功能评价方面发挥重要作用。
猜你喜欢
湖北农业科学(2022年11期)2022-07-18
河北农业大学学报(2022年2期)2022-04-26
昆明医科大学学报(2022年2期)2022-03-29
中老年保健(2021年3期)2021-12-03
实用肿瘤学杂志(2020年4期)2020-12-08
无机化学学报(2020年7期)2020-07-20
物理化学学报(2020年4期)2020-04-24
渤海大学学报(自然科学版)(2020年3期)2020-02-02
世界农药(2019年2期)2019-07-13
科技创新导报(2016年30期)2017-03-15
