
婴儿神经轴索营养不良1 例临床和基因变异分析
2021-05-01 11:21童培刘艳
临床儿科杂志 2021年4期
童 培 刘 艳
华中科技大学同济医学院附属同济医院儿科(湖北武汉 430000)
婴儿神经轴索营养不良(infantile neuroaxonaldystrophy,INAD)是一种罕见的常染色体隐性遗传的神经退行性病变[1],2006 年发现INAD 与PLA2G6基因变异有关[2]。临床表现主要以进行性智力运动倒退为特征,患儿大多于出生后3 年内起病,10岁前死亡;肌电图可有失神经电位改变;头颅磁共振成像(MRI)示小脑萎缩[3]。由于INAD在发病早期缺乏典型的临床表现,诊断较为困难。目前国内报道INAD病例少,暂无发病率相关数据。本文回顾分析1例PLA2G6基因变异所致INAD患儿的临床资料和基因检测结果,并复习国内外文献。
1 临床资料
患儿,女性,现2岁。1岁7个月时出现表情呆滞,语言和运动发育倒退就诊。患儿1 岁3 个月时会独站、扶行,逐渐进展为不能站立、不能独坐。既往可叫“妈妈”,理解简单的语言和指令,逐渐听不懂家长言语,呼名无反应。患儿发病前后均无惊厥发作。患儿系G 2 P 2,足月顺产,出生史无异常。患儿父母体健,非近亲婚配。同胞哥哥7 岁,智力和运动发育均无异常,无相关疾病家族史。体格检查:神志清楚,身长79 cm(P15),体质量9.5 kg(P15~P50),头围47.5 cm(P50~P85),双侧瞳孔等大等圆,对光反射尚灵敏,颈软,未触及肿大淋巴结,全身皮肤无黄染,无皮疹,无水肿,心肺未闻及异常,腹平软,肝脾肋下未触及,双下肢无浮肿,双下肢肌张力低,腱反射可对称引出,双侧巴氏征阳性。独坐不稳,可翻身,不会叫人及说话,不能听懂指令完成动作。实验室检查:血肝肾功能、乳酸、丙酮酸、血氨无异常;血、尿代谢筛查未见异常。MRI 示双侧小脑脑沟增宽,考虑为小脑萎缩(图1)。患儿1岁8个月时行肌电图检查示受检神经运动感觉传导大致正常,受检肌肉未见异常运动单位电位。2 h视频脑电图无异常。
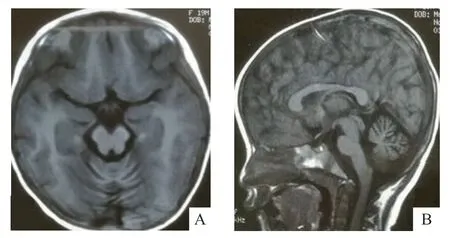
A.轴位;B.矢状位
经医学伦理审核,以及患儿家长知情同意后,抽取患儿及其父母静脉血各3 mL,提取全血基因组DNA,委托上海明码生物有限公司(CLIA 实验室ID:99 D 2064856)完成全外显子基因变异分析。对患儿基因组DNA 进行全外显子组捕获和测序,基于二代测序数据进行点变异及小片段插入缺失变异(SNV)和大片段拷贝数变异(CNV)分析发现,患儿PLA 2 G 6 基因外显子区域2 处变异:c.150_153 del移码变异和c.799 T>C 杂合点变异,分别引起氨基酸p.Pro51Thrfs*30和p.Cys267Arg变化(图2)。基因测序验证结果示2 个变异分别来源于母亲和父亲,为复合杂合变异,均未在正常人群中检测到。
2 讨论
INAD 患儿大多于3 岁前起病,最早可于新生儿期发病[4-5]。2006年研究发现,INAD与PLA2G6基因变异有关,确定其为INAD致病基因[2]。本例患儿于1岁7个月时出现智力倒退、表情呆滞,且双下肢肌张力低,腱反射活跃,双侧巴氏征阳性,与其他报道的典型INAD患儿临床表现一致。但本例患儿的肌电图未见明显异常,考虑可能和病程尚短相关。国内研究显示,21 例INAD 患儿行肌电图检查,除l 例为阴性,其余20例均为失神经支配特征,提示为神经源性损害[6]。头颅MRI 也有助于INAD 诊断,小脑萎缩伴T 2 加权高信号是INAD的典型头颅MRI特征[7]。有文献报道44例,年龄为5月龄~2岁6个月(平均年龄1岁1个月)的PLA2G6基因变异的INAD患儿,头颅MRI示小脑萎缩占95%,6 l%可见小脑高信号,48%见苍白球区异常铁沉积,14%黑质铁增多,但有2例年龄<2岁的INAD患儿头颅MRI示正常[8]。本例患儿头颅MRI示小脑萎缩,可作为支持INAD诊断的证据之一。
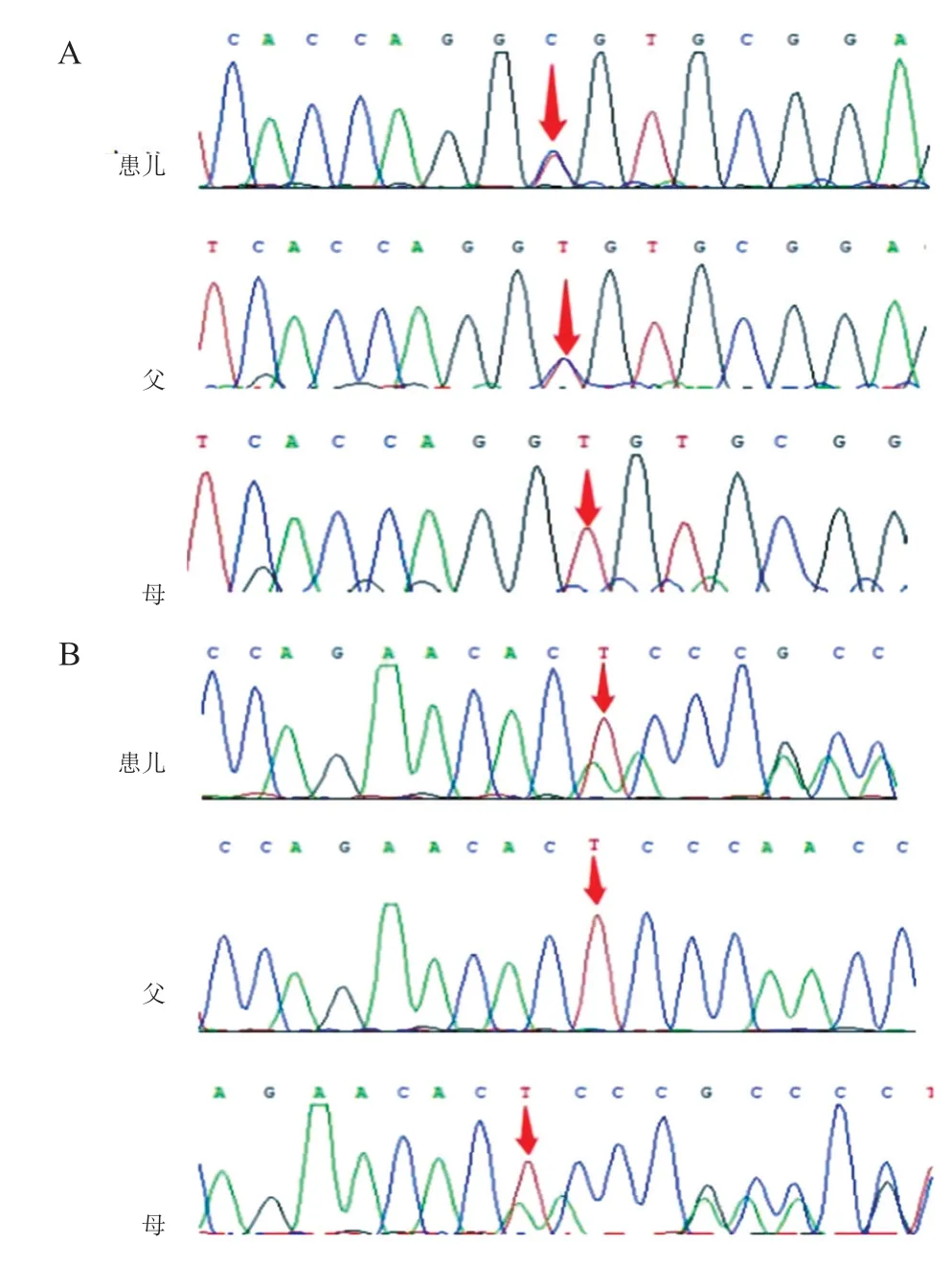
图2 患儿及其父母PLA2G6 基因Sanger 测序图
检索中国知网、万方、PubMed等数据库,近10余年国内外已报道200余例INAD患儿。国内报道的37例INAD 患儿均有智力运动发育倒退,其中4 例有惊厥发作,3例有眼球震颤等;除3例患儿未检测到任何基因变异外,其余均为PLA2G6基因变异[6,9-14]。
PLA2G6基因位于第22号常染色体,6 Mb,有17个外显子,编码氨基酸806个,产物为细胞质内非Ca2+依赖的第Ⅵ组磷脂酶A2,其活性单位为四聚体结构。磷脂酶A2Ⅵ蛋白在磷脂改造、花生四烯酸的释放、白三烯和前列腺素的合成与调亡中具有重要作用[15]。iPLA2-VIA属于PLA2超家族成员,能水解磷脂中的sn-2 酯键,并产生游离脂肪酸和溶血磷脂,若蛋白质发生功能缺失则可引起脂质过氧化反应、线粒体内膜损伤、细胞内铁沉积,高尔基体形态改变,最终会导致细胞凋亡以及神经退行性变。PLA2G6基因变异也可引起酶(包括磷脂酶和溶血磷脂酶)活性丧失,并且不能催化释放脂肪酸,从而引起磷脂底物堆积,最终导致神经轴索变性。
目前已发现的PLA 2 G 6 基因变异大多分布于PLA2G6蛋白N端至C端的各个结构域中,其中绝大部分为错义变异,小部分为无义变异、拷贝数变异及剪接位点变异[16]。国内文献报道的37例INAD患儿,PLA2G6基因变异亦以错义变异为主。基因检测显示本例患儿PLA2G6基因区域2处变异:c.150_153del移码变异和c.799 T>C 错义变异,分别引起氨基酸p.Pro51Thrfs*30和p.Cys267Arg发生变化;尽管蛋白质功能预测提示来自父亲的错义变异c.799T>C导致蛋白质功能损害,但来自母亲的移码变异,理论上可导致该基因蛋白翻译提前终止,该变异下游有多个无效变异致病的报道,提示蛋白缺失部分对蛋白功能仍有重要影响。研究表明,PLA 2 G 6 基因缺失后,线粒体功能可出现异常,包括线粒体ATP 合成障碍、线粒体形态发生变化、呼吸链功能降低[17],这些异常表明PLA2G6基因异常会使机体氧化应激反应增强,从而影响机体正常的新陈代谢。目前尚无研究表明INAD患儿临床症状轻重与基因变异类型具有相关性。
对于INAD 的治疗,目前国内多采取对症支持治疗,并无针对该基因的特殊用药。研究显示,INAD可采用酶替代、基因替代以及基因校正等治疗方法,其治疗效果有待进一步研究[18]。多数INAD患儿预后较差,于10岁前死亡[19]。
猜你喜欢
分子诊断与治疗杂志(2022年9期)2022-10-09
保健与生活(2022年9期)2022-05-06
心电与循环(2021年4期)2021-11-29
健康博览(2021年7期)2021-08-16
保健与生活(2020年11期)2020-06-23
家庭科学·新健康(2016年9期)2016-10-25
数理化学习·高三版(2015年3期)2015-10-21
湖北农业科学(2014年11期)2014-09-10
职业·中旬(2009年12期)2009-06-01
