
COA6 基因新发变异致婴儿细胞色素 c 氧化酶缺陷致心脑肌病4 型 1 例报告并文献复习
2021-05-01 11:20崔清洋曹银利唐成和石计朋
临床儿科杂志 2021年4期
崔清洋 尚 云 曹银利 唐成和 石计朋
新乡医学院第一附属医院儿科(河南卫辉 453100)
基因变异干扰线粒体呼吸链(mitochondrial respiratory chain,MRC)功能所致的线粒体呼吸链疾病(MRC disease,MRCD)是一种最常见的先天性疾病之一,发病率估计为1:5000活产儿。MRCD为基因异质性疾病,可由线粒体和/或核基因变异引起。已知的MRC缺陷已经扩展到110个基因,且随着测序技术的进步,这个数值还在快速增长。
复合体Ⅳ(细胞色素 c 氧化酶,cytochrome c oxidase,CcO)是呼吸链的末端酶,负责把电子从细胞色素c转移到分子氧,由13个多肽亚基组成,其中3个亚基由线粒体基因编码,其他10个为核基因编码。细胞色素氧化酶复合体中的3种线粒体编码蛋白质是执行电子传递功能的实际催化亚单位。可导致CcO缺陷者包括核编码的基因和线粒体编码的基因,其中线粒体编码基因有MT-CO1、MT-CO2、MT-CO3、MT-TS1及MT-TL1,核编码基因有COX10、COX6B1、SCO1、C 12 ORF 62、COX 20、APOPT 1、COA 3、COX 8 A、COX 6 A 2、COX 4 I 1、COXFA 4、PET 117 和COX 5 A。而与致死性婴儿心脑肌病尤为关联的基因为 SCO2、COX15、COA5和COA6。
婴儿细胞色素 c 氧化酶缺陷致心脑肌病4 型(infantile cardioencephalomyopathy due to cytochrome c oxidase deficiency-4,CEMCOX4)是一种呈常染色体隐性遗传的线粒体疾病,由常染色体1q42上的COA6基因纯合或复合杂合变异所致,其特征是在宫内或出生后的前几天发病,临床特征为心肌病、脑肌病、骨骼肌肌病、Leigh综合征、代谢性酸中毒和偶发的肝衰竭;此外大多数患儿还表现出神经系统异常,如呼吸异常、眼球震颤和脑回异常,均符合脑病。这种疾病通常在婴儿早期是致命的。目前尚无CEMCOX4的发病率报道,国际上均为个案报道,国内尚无报告。本文回顾分析1例CEMCOX4患儿的临床资料及基因检测结果,并首次报道COA6基因c.411_412insAAAG纯合变异。
1 临床资料
女性患儿,5 日龄,因呼吸困难5 天、加重6 小时入院。患儿于出生后不久即出现呼吸困难,无发热及呻吟,无腹部平坦或凹陷,就诊于当地医院予经鼻持续气道正压通气(nasal continuous positive airway pressure,nCPAP)及抗感染治疗4 天后呼吸困难减轻,予鼻导管吸氧后经皮血氧饱和度尚稳定。入院前6 小时患儿呼吸困难再次加重,予气管插管及呼吸机辅助通气下转入新乡医学院第一附属医院。患儿系G 1 P 1,足月顺产,无产伤及窒息史。父母均体健,非近亲结婚。体格检查:呼吸45 次/min,心率无,予心肺复苏后心率恢复正常,一般情况及反应差,肢体少动,面色青灰,全身皮肤发花,肢体末端凉,皮肤弹性稍差,双侧瞳孔对光反射迟钝,气管插管下呼吸费力,吸气三凹征阳性,双肺未闻及明显啰音,心音亢进,心前区可闻及Ⅲ/6 收缩期吹风样杂音,肝脏肋下4 cm,质软,肠鸣音未闻及,四肢肌张力低下,原始反射未引出。实验室检查:血气分析示代谢性酸中毒及高乳酸血症,碳酸氢钠治疗后多次复查均提示高乳酸血症;血常规白细胞16.5×109/L,红细胞 4.13 × 1012/L,血红蛋白158 g/L,血小板128×109/L;大小便常规未见异常;肝功能、电解质未见异常;尿素13.73 mmol/L,尿酸408 μmol/L,肌酐85.4 μmol/L;乳酸脱氢酶911 U/L,α-羟丁酸脱氢酶605 U/L,肌酸激酶2 928 U/L,肌酸激酶同工酶215 U/L;B型钠尿肽前体36 000 pg/mL;纤维蛋白原80.7 mg/dL;淋巴细胞亚群未见异常;IgG 9.3 g/L,IgA 0.02 g/L,IgM 0.05 g/L;血氨75 μmol/L,乳酸12.0 mmol/L;超敏C 反应蛋白12.8 mg/L;血培养人葡萄球菌生长。胸片示心影大,右肺纹理模糊。心脏彩超考虑肥厚型心肌病,右心室及左心室流出道梗阻,冠状动脉略增宽,卵圆孔未闭,动脉导管未闭。血串联质谱(不同时间2次)示多种中长链酰基肉碱增高。尿气相色谱-质谱示4-羟基苯乳酸增高,可能继发于肝损伤。入院诊断:新生儿肺炎并呼吸衰竭,心力衰竭,肥厚型心肌病,新生儿败血症,高乳酸血症,多种酰基辅酶A 脱氢酶缺乏症?患儿入院后予抗感染、呼吸机辅助通气、抗心力衰竭、降低心率减轻心室流出道梗阻、静脉营养、补充维生素B2及左卡尼丁等治疗后病情逐渐好转,撤离呼吸机后自主呼吸尚可,但复查感染指标反复,升级抗生素后复查感染指标逐渐恢复正常。
因患儿多次血气分析提示高乳酸血症,血串联质谱提示多种酰基辅酶A脱氢酶缺乏症可能,在家属知情同意后行基因检测。提取患儿及其父母外周血基因组 DNA,使用 Illumina 高通量测序仪和 Agilgent 公司的 SureSelect 探针富集体系进行二代基因测序。使用软件 CASAVA(1.8.2)将原始数据转化为可识别的碱基序列,然后进行 Align分析、SNP分析和DIP分析,获得靶向区域变异位点的信息。通过 PolyPhen-2、SIFT、Mutation Taster 进行蛋白质损伤分析,获得需要进一步验证的变异位点。在人类基因组数据库GenBank 中获得 COA 6 基因变异位点基因序列,在引物设计网站 Primer Z(http://genepipe.ncgm.sinica.edu.tw/primerz/primerz 4.do)设计并合成引物。对变异位点进行 PCR 扩增后进行一代测序验证,排除掉二代测序中假阳性的位点。NGS 测序分析发现患儿COA6 基因第3外显子c.411_412insAAAG纯合移码变异(p.Lys140 Arg fs Ter4),从第140个赖氨酸开始的氨基酸合成发生改变,并在改变后的第 4 个氨基酸终止。c.411_412insAAAG变异可能造成蛋白质的功能受到影响,c.411_412insAAAG变异尚未见文献报道(参考数据库 HGMD Pro及 PubMed)。该变异不归类为多态性变化,在人群中的发生频率极为低下(参考数据库 1000 Genomes 和dbSNP)。经分析未见可疑的 CNV 变异。线粒体基因二代测序检测未发现受检者有临床意义的线粒体基因变异;MLPA 检测未发现受检者线粒体基因存在大片段变异。家系验证显示,父母均为COA 6 基因c.411_412 insAAAG 杂合携带者,符合常染色体隐性遗传规律(图 1)。但未发现ACADVL、ETFA、ETFB 或ETFDH 基因变异。根据美国医学遗传学与基因组学会(ACMG)联合美国分子病理学会(AMP) 2015 年制订的“基因序列变异解释的标准和指南”进行致病性分析[1]。COA 6 基因c.411_412 insAAAG 的致病性:①当某基因致病机制为功能缺失(loss-of-function,LOF)时,该基因上的无义变异、移码变异、±1 或 2 位置的剪切变异、起始密码子变异、单个或多个外显子缺失(非常强的致病性证据,PVS 1);②c.193 delC 变异通过比对千人基因组数据库(1000 Genomes)、人类基因突变数据库(HGMD)未见收录(中等致病性证据,PM 2)。综合上述c.411_412 insAAAG 变异的证据强度为“PVS 1+PM 2”,判断为造成患儿发病的疑似致病性变异。
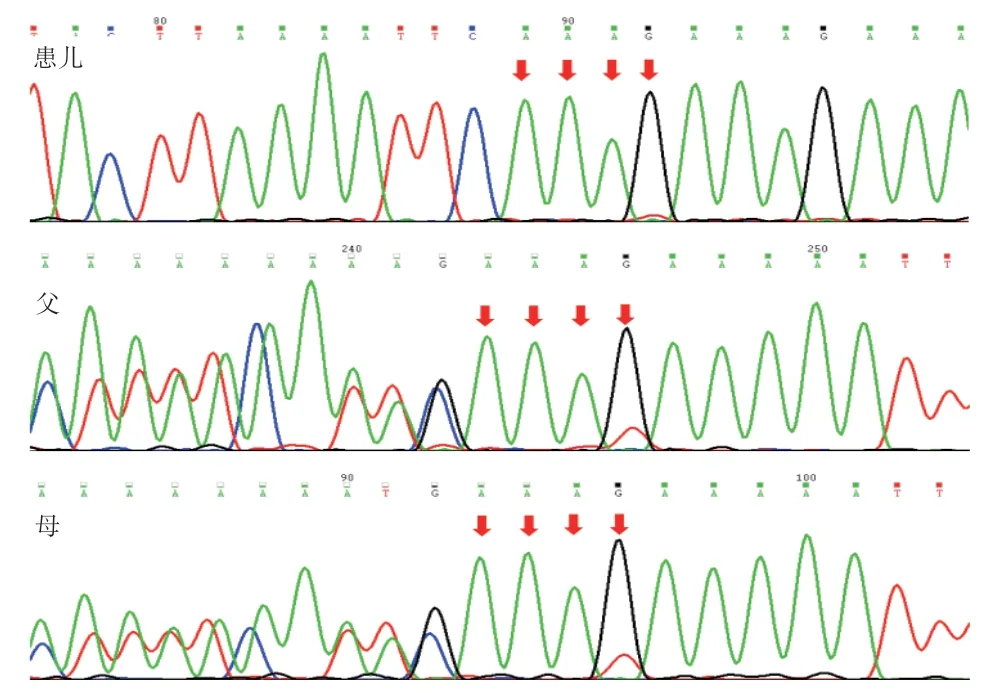
图1 患儿及父母COA6 基因c.411_412 ins AAAG 变异测序图
结合患儿临床表现、心脏彩超及基因检测结果,诊断为COA6基因c.411_412 ins AAAG纯合变异所致的CEMCOX4。患儿出院后继续口服美托洛尔、维生素B2和左卡尼汀,告知家属患儿有猝死风险,勿烦躁哭闹。定期复查乳酸显示较住院时下降,但仍未恢复至正常范围,停用维生素B2和左卡尼汀。
2 讨论
COA6基因定位于1q42,基因组全长约10.59 kb,包含3个外显子和2个内含子,外显子长度约710 bp,编码155 个氨基酸。目前尚无COA6 基因变异以何种变异为最多的报道,也无基因型与表型关系的报道。
CcO 是呼吸链的末端酶且是细胞呼吸的主要部位。CcO 是进化上高度保守的多亚基酶复合体,由酵母中的11 个亚基和哺乳动物中的13 个亚基组成。除了蛋白质亚基外,CcO 还包括几个辅因子,包括2 个Cu 位点(CuA 和CuB),2 个血红素基团(血红素a 和a3)和1个镁和锌离子。细胞色素c锚定在COX2的膜间隙上,它包含1个双核铜中心,称为CuA,接受电子,之后电子被传递至COX1中的血红素基团,再传递至血红素a3-CuB位点,最后传递至分子氧,后者被还原成水。CcO 的生物合成过程极其复杂,需要最多达40种不同的组装因子。Coa6是一种具有CX9C-CX10C序列基序的可溶性膜间隙蛋白,人类细胞中Coa 6 缺失导致COX组装和活性降低,但这种缺陷可以通过外源性的铜补充来部分挽救[2]。
CcO的组装缺陷具有临床异质性,可累及机体的1个或多个器官系统,发病年龄从婴儿到成年期不等。这些疾病(线粒体复合体Ⅳ缺陷)以广泛的临床表型为特征,包括心肌病、脑肌病、骨骼肌肌病、Leigh 综合征、代谢性酸中毒和偶发肝衰竭。但只有少数复合体Ⅳ可追踪到CcO的核心亚基,且大多数患儿在编码组装因子的基因中发现了变异,其中包括富含亮氨酸的五肽重复序列即motif-containing蛋白LRPPRC——一种线粒体mRNA稳定蛋白,TACO1——一种COX1的翻译激活剂以及人类COX14同源物,C12orf62——一种参与COX 1 合成和组装的因子;同样在COX 10和COX 15、血红素合成所需CcO 组装因子以及将铜插入CcO 所需的SCO 1 和SCO 2 基因中也发现了致病性变异。但其他疾病基因,包括SURF1、C2orf64/COA5和FAM36A在CcO组装中的确切作用尚未完全明确。
有报道1 例男婴罹患因COA 6 基因复合杂合变异所致的致命性婴儿心脑肌病,6 月龄时诊断为肥厚型梗阻性心肌病,且有肌肉体积下降和肌张力降低以及进行性无力和嗜睡,18 月龄死于心力衰竭[3-4]。对酵母同源物的研究表明,变异导致Coa 6 的线粒体水平下降,变异蛋白无法补救Coa 6 缺失酵母的线粒体呼吸生长缺陷,与功能丧失一致。Morpholino敲除斑马鱼胚胎COA 6 基因可导致心脏发育和功能缺陷。
另有报道1例阿拉伯血统罹患因COA6基因变异所致的致命性心脑肌病女婴,其父母近亲婚配;出生时患儿表现为肌张力降低、心前区收缩期杂音及轻度的畸形特征(面容异常,包括小下巴及眼眶扁平),出生后不久进展为严重的乳酸酸中毒、体温过低和呼吸急促;超声心动图显示影响双侧心室的严重肥厚型心肌病,伴左心室部分区域致密化不全,二尖瓣、三尖瓣和肺动脉瓣关闭不全;5 周龄时去世。经全外显子组测序及Sanger验证,在患儿的COA6基因中发现纯合错义变异(W66 R)。患儿成纤维细胞COA6蛋白缺乏和线粒体复合体Ⅳ减少,用铜处理成纤维细胞可使复合体Ⅳ及其亚基的稳定性增加,提示可能的治疗选择[5]。
婴儿CcO缺陷致心脑肌病在遗传上具有异质性,除了染色体 1q42上的COA6基因可造成婴儿CcO缺陷致心脑肌病4型外,尚有染色体10q24上的COX15基因变异所致的CEMCOX2、染色体 2q11上的COA5基因变异所致的CEMCOX3。
与致死性婴儿心脑肌病关联密切的基因有SCO2、COX15、COA5和COA6。希腊和美国联合报道了3例无血缘关系的SCO2基因变异引起致死性婴儿心脑肌病,均表现为肥厚性心肌病、乳酸性酸中毒和胶质增生。心脏和骨骼肌活检显示COX活性降低,而肝脏和成纤维细胞有轻度COX 缺乏。其中2 例患儿的肌肉组织化学显示在所有纤维中COX 酶活性均降低,但琥珀酸脱氢酶活性正常,未见碎红纤维。免疫组织化学显示线粒体编码的COX Ⅰ和COXⅡ亚基严重减少,而核编码的COX Ⅳ亚基和COX Ⅴa 存在,但活性下降。由SCO 2 基因变异引起的COX 缺乏症的临床表型与其他基因变异引起的临床表型有所不同。德国报道了来自2 个无血缘关系家庭的3 例SCO 2 基因变异引起致死性婴儿心脑肌病患者,他们出生时出现低血压、呼吸困难、血液和脑脊液乳酸增加、肥厚性心肌病和严重COX 活性下降,均在婴儿期死亡。美国报道了1 名妇女,第1 次怀孕在妊娠11 周自然流产;第2 个孩子于53 天死于致死性婴儿心脑肌病,该患儿SCO 2 基因有2 个复合杂合变异;第3 次怀孕也在妊娠10周自然流产,流产物基因检测显示与第2胎相同SCO 2 基因变异,SCO 2 基因变异可能与早期自然流产有关。荷兰也报道2例同胞胎罹患致死性婴儿心脑肌病,先证者出生时四肢肌张力低下和呼吸功能不全,之后进展为癫痫发作和吸气性喘鸣(认为部分原因与脑干异常有关),出生后第25 天死于肥厚性心肌病;实验室检查显示乳酸和丙酮酸增高,心脏彩超检查显示进行性肥厚性心肌病,尸检显示心肌细胞胞浆空泡化,脑显像示与线粒体疾病相符的脑室周围白质、皮质脊髓束和胼胝体改变;心脏(正常对照值的1%)、肌肉(19%)和培养的成纤维细胞(16%)COX-Ⅳ活性降低;遗传学分析确定了SCO 2 基因2个复合杂合变异(E 140 K 和W 36 X);在其第2 胎妊娠中期进行的羊膜穿刺基因检测时发现携带了SCO2基因相同的变异,遂于妊娠23周选择人工流产,流产物尸检显示女性胎儿心脏明显增大和脑回复杂程度降低,提示产前心脏和大脑的异常。有报道1 例出生时持续性呼吸窘迫需要呼吸支持的小头畸形女婴,出生不久即有持续性乳酸酸中毒脑病,出生第9 天持续出现脑病,伴运动减弱及呼吸抑制,拔除气管插管20分钟后死亡;头颅MRI显示细微改变,内囊后肢没有明显髓鞘形成;心脏彩超显示所有心肌壁明显肥大及射血分数降低;死后骨骼肌活检显示复合体Ⅳ活性轻微降低,而心肌中却发现明显的复合体Ⅳ缺乏,其他呼吸链复合体活性不显著;病理组织学显示心脏双心室肥大,伴有糖原沉积及COX 染色缺乏,此外骨骼肌肌原纤维间质和浆膜下糖原增多,肝脏脂肪变性,脑胶质增生;基因测序提示COX 15 基因父源性c.452 C>G 及母源性c.649 C>T 复合杂合变异[6]。另有报道,1对父母为土耳其血统的双胎兄妹,其CcO缺乏表现为致命性新生儿肥厚性心肌病,分别在出生第8天和第10天死亡;尸检显示心肌细胞内脂肪滴聚集,线粒体增殖;但2 例患儿均无大脑或骨骼肌损害的记录;成纤维细胞染色提示复合体Ⅳ活性严重下降;基因测序显示COA5基因纯合c.157G>C变异[7]。
上述文献复习显示,无论是COA 6 基因,还是SCO2、COX15、COA5基因所致的致死性婴儿CcO缺陷致心脑肌病临床多表现为乳酸酸中毒、肌张力低下及肥厚性心肌病,大多预后极差。本例患儿出生后持续呼吸困难5 天,检查提示多次高乳酸血症和肥厚性心肌病,虽2 次血串联质谱均提示多种酰基辅酶A脱氢酶缺乏症可能,且补充维生素B2及左卡尼汀后乳酸稍有下降,但仍未降至正常范围,经全外显子及线粒体基因检测最终诊断为 CEMCOX 4。尽管依据ACMG和AMP“基因序列变异的解释标准和指南”进行的致病性分析尚不能确认COA 6 基因c.411_412 insAAAG纯和变异位点的致病性,但患儿有较为明显的CEMCOX4临床表现,且c.411_412 insAAAG纯和变异不属于多态性变化,在人群中出现的概率极其低下(参考的数据库为1000 Genomes 及dbSNP),综合患儿的基因检测结果及临床表型,可基本确认c.411_412 insAAAG 纯和变异为患儿的致病性变异。
本研究报告1例CEMCOX4 病例,其COA6 基因c.411_412 ins AAAG纯合变异国际上尚未见报道,扩充了CEMCOX4的基因变异谱。
猜你喜欢
农业工程学报(2022年13期)2022-10-09
大气科学学报(2022年2期)2022-05-12
中国典型病例大全(2022年9期)2022-04-19
聊城大学学报(自然科学版)(2022年3期)2022-02-14
作物学报(2022年3期)2022-01-22
食品安全导刊(2021年33期)2021-11-27
阅读(科学探秘)(2020年9期)2020-11-06
保健与生活(2020年13期)2020-07-24
家庭医学·下半月(2019年5期)2019-07-12
家庭医学(2019年2期)2019-06-10
