
CACNA1E 基因变异致儿童难治性癫痫1 例报告
2021-05-01 11:20李洁玲
临床儿科杂志 2021年4期
李洁玲 曹 洁
重庆医科大学附属儿童医院内科全科 儿童发育疾病研究教育部重点实验室 国家儿童健康与疾病临床医学研究中心 儿童发育重大疾病国家国际科技合作基地 儿科学重庆市重点实验室(重庆 400014)
癫痫是儿童常见的神经系统疾病之一,其中约20%~30%的患儿为难治性癫痫,难治性癫痫出现反复癫痫发作,对抗癫痫药物疗效差,难以控制,严重的可以伴有智力运动发育落后及癫痫脑病。难治性癫痫的病因繁多,甚至大部分病因尚不完全明确。随着高通量测序技术的不断发展,越来越多的研究发现难治性癫痫与许多基因变异可能有关。本文结合临床案例,主要探讨CACNA1E基因变异所致难治性癫痫的临床特点及诊治情况,为该疾病的临床诊断与精准治疗提供参考。
1 临床资料
患儿系G2P2,39+2周,剖宫产,Apgar评分10-10-10。出生体质量3 380 g,羊水性状正常,量偏多,脐带绕颈1圈,胎盘无异常,无宫内窘迫、胎膜早破,无围生期缺氧窒息抢救史,母亲孕期无异常,父母体健,非近亲婚配,家族中无惊厥及癫痫病史。患儿生后不吃、不哭、不动、反应差,偶有尖叫、肢体抖动,无双目凝视,无意识障碍。出生时体格检查:体质量3 330 g,头围37 cm,身长51 cm,成熟儿外貌,营养可,反应差,刺激不哭(有皱眉),前囟平软,双侧腕关节外展,手指细长呈爪型,掌纹异常,双侧踝关节内收,足趾内收、重叠,四肢肌张力减弱,原始反射未引出。实验室检查:血气分析、血氨、血乳酸、新生儿代谢性疾病筛查、血串联质谱、尿有机酸、高分辨染色体检查均无异常。脑电图无异常。头颅磁共振成像(MRI)示双侧苍白球T1信号对称性偏高。
患儿3 月龄时反复出现频繁双眼上翻或眼球转动,脑电图示变异性高幅失律,临床诊断婴儿痉挛症,先后予左乙拉西坦、托吡酯、氯硝西泮、丙戊酸钠口服抗癫痫治疗,但效果不佳,出现成串痉挛发作(屈曲型),每天发作数十次不等,最多达到100余次。
患儿于11 月龄、1 岁3 个月时因频繁惊厥发作不易控制先后2次入院治疗。患儿目前1岁4个月,智力运动发育较同龄儿明显落后,不能追光、追物,不能抬头,不能坐,不能逗笑,不能认人,只能发音。体格检查:发育迟滞,营养中等,嗜睡,反应差,面色欠佳,高腭弓,双手通贯掌,四肢肌力降低,肌张力降低,双上肢可见轻微自主活动,双下肢不能自主活动。多次脑电图提示变异性高幅失律(图1),头颅CT 示脑萎缩(图2)。入院后促肾上腺皮质激素静滴14天(50 U/d)治疗,发作基本控制。脑电图改善。逐渐改为泼尼松口服,1个月后再次反复痉挛发作,脑电图仍呈变异型高幅失律。又进行生酮饮食治疗,患儿在生酮饮食过程中,反复出现低蛋白血症、低钾血症,无法耐受,停用生酮饮食治疗,加用氯巴占、唑尼沙胺口服,患儿癫痫发作情况较前有好转,但每日仍有数次至数十次惊厥发作不等,主要表现为扭动脖子、耸肩、双手握拳伴眼球转动。
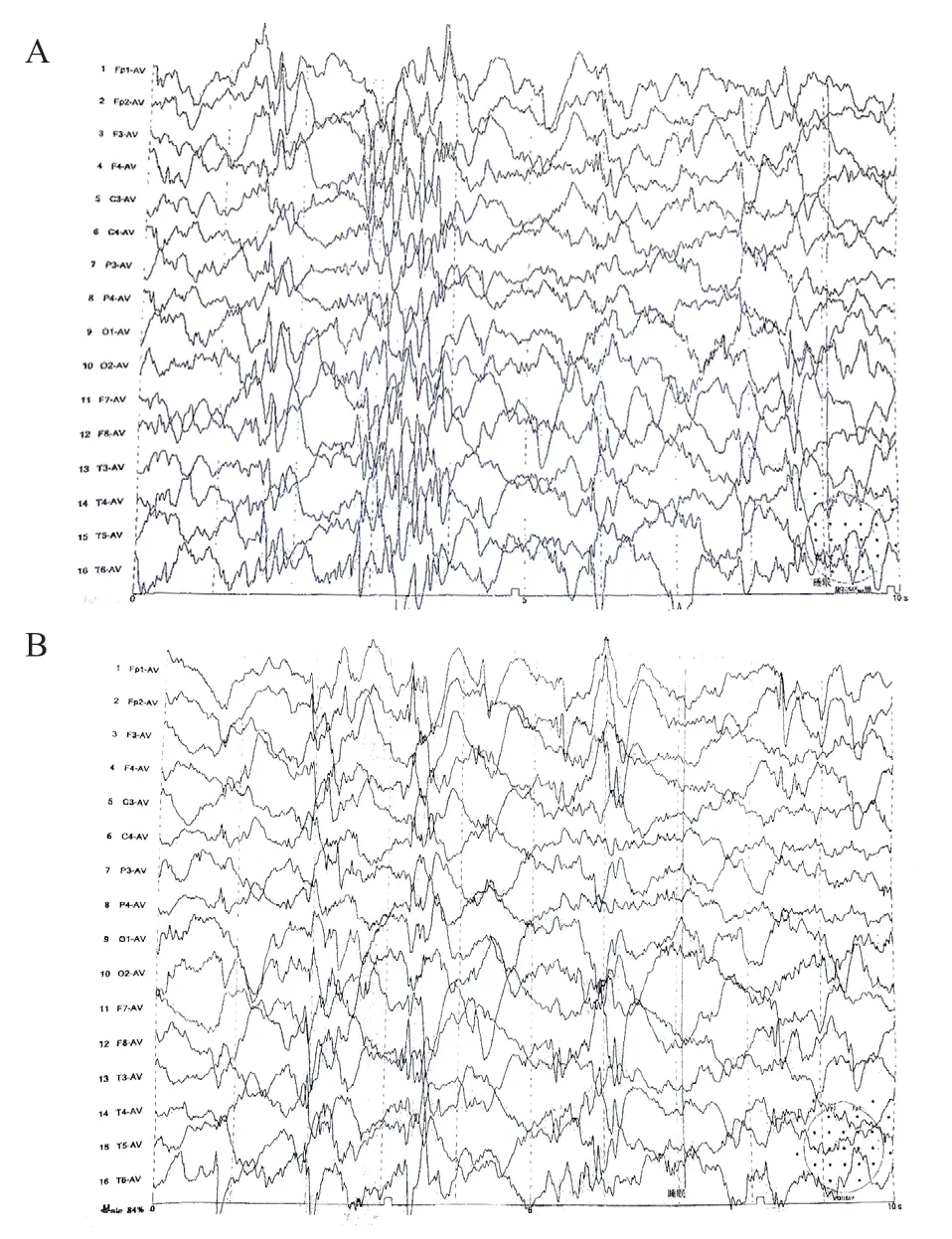
图1 患儿脑电图表现
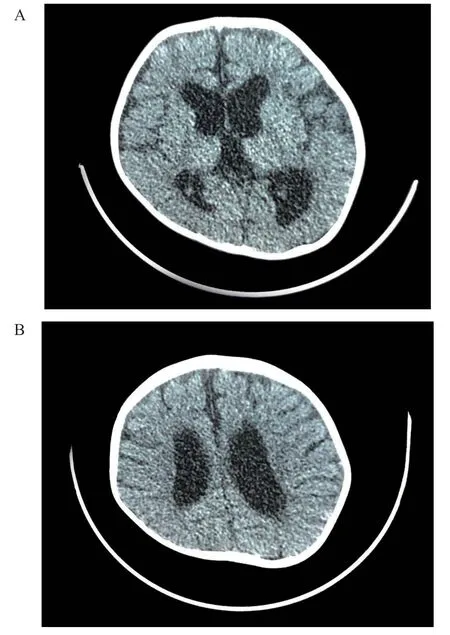
图2 患儿头颅CT 表现
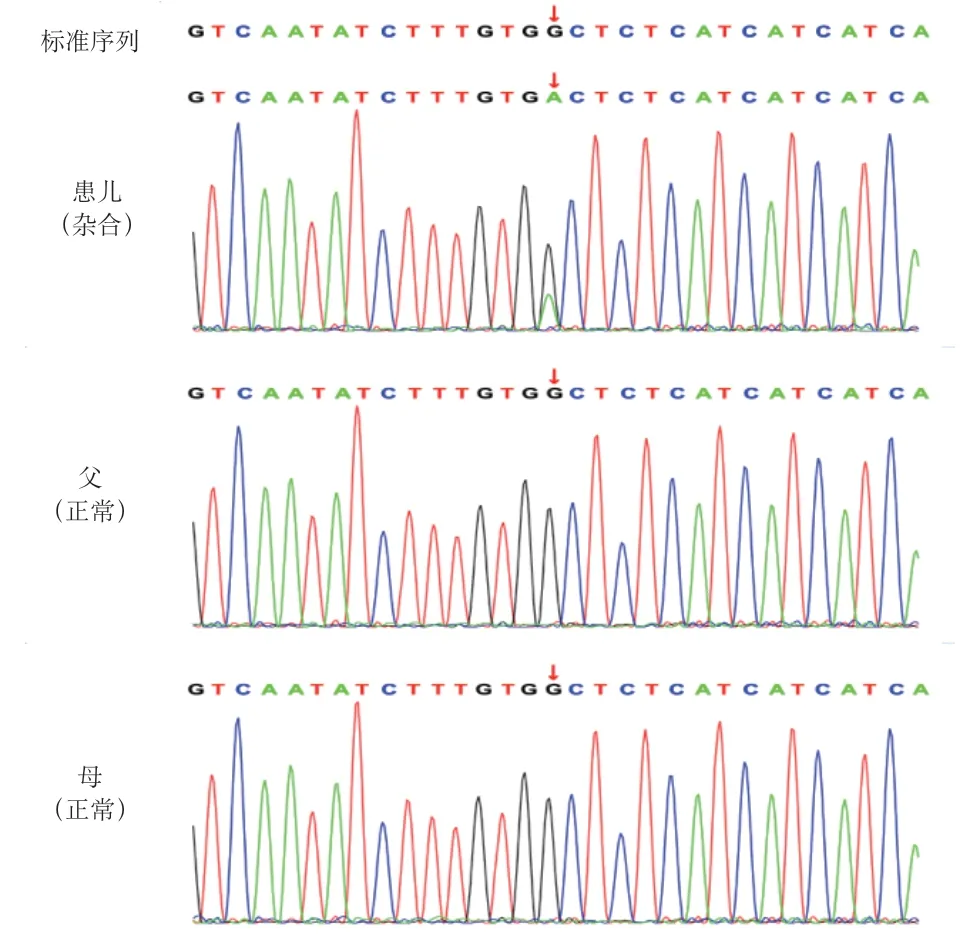
图3 患儿CACNA1E 基因Sanger 测序
鉴于该患儿反复癫痫发作较严重,且原因不明,同时伴有运动智力发育明显落后及严重癫痫脑病表现。经医学伦理审核,患儿父母知情同意后进行基因检测。抽取患儿及父母各2 mL 血进行医学全外显子检测,检测分为变异筛查(运用高通量测序技术)、基因数据分析(运用生物信息学及临床信息分析技术)和疑似致病变异验证(运用Sanger 测序技术)3 个主要步骤。基因检测结果提示,患儿携带CACNA 1 E 基因c.4258(exon 30)G>A(NM_001205293)新发杂合变异,该位点变异引起蛋白产物第1420 位氨基酸错义变异(p.A 1420 T),而患儿父母该位点野生型,家系Sanger 验证结果见图3。CACNA 1 E 基因c.4258(exon 30)G>A 变异,在各频率数据库(dbSNP,千人基因组,EXAC)中均无报道,属于低频变异,而且多种统计方法预测出该变异对基因(基因产物)有影响:保守性及蛋白结构预测软件[sift(damaging),Polyphen2_HDIV(probably damaging),Polyphen2_HVAR(probably damaging),PROVEAN(deleteriou),MutationTaster(disease_causing),M-CAP(damaging),REVEL(deleterious),GERP(D),phyloP20way(D),phastCons 20 way(D)]预测出该变异对基因(基因产物)有影响。根据ACMG(The American College of Medical Genetics and Genomics)标准与指南(2015),患儿CACNA 1 E 基因c.4258(exon 30)G>A(NM_001205293)变异符合证据PS 2+PM 2+PP 3,最终评定为可能致病性变异。结合基因检测结果,患儿诊断为:癫痫,癫痫性脑病(CACNA 1 E 基因相关)。
患儿目前继续口服丙戊酸钠、左乙拉西坦、托吡酯、氯巴占、唑尼沙胺,但仍有反复惊厥发作,发作频率无明显改善。现反复调整抗癫痫药物,其中托吡酯减量后,患儿癫痫发作频繁,重新加量后,癫痫能得到一定控制。
2 讨论
在哺乳动物中枢神经系统中,电压门控钙通道控制钙离子的内流,形成膜去极化,从而调节细胞内信号和基因表达。α 亚单位是钙通道的主要亚单位,由10 个基因编码,包括CACNA 1 A、CACNA 1 B、CACNA 1 C、CACNA 1 D、CACNA 1 E、CACNA 1 F、CACNA 1 G、CACNA 1 H、CACNA 1 I、CACNA 1 S[1]。以上这些基因在特异的细胞和组织中表达,其中许多与人类疾病有关,其中有研究报道CACNA 1 A、CACNA1D、CACNA1E、CACNA1G变异与癫痫有关[2-4]。本例患儿为CACNA1E基因变异引起的难治性癫痫。
CACNA 1 E 基因编码具有高压激活、快速失活功能的R 型钙通道亚单位,这在中枢神经系统中启动快速突触传递[5]。以小鼠CACNA 1 E 为探针,从海马互补DNA文库中克隆CACNA1E,为编码2 251个氨基酸组成的蛋白质,相对分子质量约为255 000;RTPCR 结果显示,所有被测神经元组织均有表达,在人肾、神经母细胞瘤和小鼠视网膜、垂体前叶、脾脏和胰岛细胞,以及大鼠胰岛素瘤细胞株中也检测到这种表达,同时小鼠脑切片原位杂交也显示了广泛的表达[6]。由此可见,CACNA 1 E 在中枢神经系统中起着重要作用,其发生异常,可导致神经系统紊乱,从而引发相关疾病。
早期婴儿癫痫性脑病-69(early infantile epileptic encephalopathy-69,EIEE 69)是由染色体1 q 25.3 上CACNA1E基因杂合变异引起的。有研究报道30例有相似神经系统发育障碍的无血缘关系患者,其主要临床特征包括早期发作的难治性癫痫,严重的张力低下以及智力运动发育落后;大多数患者在生后前几个月就有不同程度的癫痫发作,其中6 例在生后第一年死亡,部分在癫痫发作后出现智力运动发育倒退;其他常见的临床表现还包括痉挛性四肢瘫痪、反射亢进、运动亢进、肌张力障碍、肌阵挛、阵挛、眼神接触不良或缺失、眼球震颤、皮质视觉障碍等[5]。因此,EIEE69是一种常染色体显性遗传的严重神经发育性脑病,其特征是早期难治性癫痫发作、肌张力低下和严重发育障碍,常伴有巨头畸形、运动亢进和挛缩,可导致早期夭折,托吡酯可能对部分患者有良好的抗癫痫效果。
本例患儿有早期难治性癫痫发作、肌张力低下和严重的智力运动发育落后,与上述文献报道的EIEE69主要临床特征相符合,而且本例患儿基因检测结果显示存在CACNA 1 E 基因的杂合错义变异c.4258(exon 30)G >A,为新发变异,该变异导致氨基酸改变(p.Ala1420Thr,NM_001205293),在1420位氨基酸由丙氨酸变异为苏氨酸,从而推测可致病。因此本例患儿符合CACNA 1 E 基因变异引起的EIEE 69。在治疗过程中,托吡酯减量,患儿癫痫发作即频繁,重新加量后,癫痫能得到一定控制。由此可见,托吡酯在CACNA 1 E 基因变异导致的难治性癫痫中抗癫痫作用非常重要,这可能与托吡酯能阻断R 型钙通道有关。
综上所述,对有难治性癫痫、智力运动发育落后、严重癫痫脑病、肌张力低下等表现的患儿,应尽早考虑完善基因检测明确致病原因,以利于进一步指导治疗及判断预后。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
基层中医药(2022年1期)2022-07-22
中国典型病例大全(2022年10期)2022-05-10
中国典型病例大全(2022年10期)2022-05-10
中国康复(2021年6期)2021-11-30
中老年保健(2021年5期)2021-08-24
中老年保健(2021年6期)2021-08-24
中老年保健(2021年11期)2021-08-22
家庭百事通·健康一点通(2017年8期)2017-08-18
中国医学创新(2016年33期)2017-02-28
