
PPARγ 基因过表达/沉默对缺氧诱导的肾小管上皮细胞坏死性凋亡标志物表达调控作用
2021-03-27 11:48蹇淑娟涂立邹家森朱仕群覃远汉
山东医药 2021年6期
蹇淑娟,涂立,邹家森,朱仕群,覃远汉
广西医科大学第一附属医院,广西南宁530021
肾间质纤维化(RIF)被认为是慢性肾脏疾病发展为肾衰竭的最常见途径,是各类肾脏疾病进展至终末期肾病的必然结果,目前其发病机制尚未完全明确。研究[1]提示,肾小管上皮细胞(RTEC)损伤可能是RIF 发生发展的关键因素和中心环节,其中缺氧为RTEC 损伤的重要原因之一,可导致坏死性凋亡的发生。坏死性凋亡是一种不依赖caspase 机制、兼有坏死和凋亡双重特征的程序性坏死,它的发生主要取决于丝氨酸-苏氨酸激酶3(RIPK3)的激活[2]。研究[3]显示,RIPK1 在核心信号转导级联中也发挥了重要的作用。受体交互作用蛋白1(RIP1)和RIP3作为坏死性凋亡的上游分子信号,当坏死性凋亡启动后,RIP1 发生泛素化、去泛素化、磷酸化等一系列反应;而当蛋白激酶8 被抑制时,RIP1 与RIP3 发生相互磷酸化,形成坏死性凋亡小体,从而介导坏死性凋亡的发生[4]。因此,RIP1、RIP3不仅是坏死性凋亡发生发展的关键信号,也是坏死性凋亡的标志性蛋白。过氧化物酶体增殖物激活受体(PPARs)是一类核转录因子,属Ⅱ型核激素受体超家族成员,根据编码基因的不同可分为三种细胞亚型,分别为PPARα、PPARβ 和PPARγ,其中PPARγ 参与了正常肾脏的发育、脂质代谢,具有调节水盐重吸收和调控肾血流量并激活肾素血管紧张素系统等生理功能。研究[5]显示,转化生长因子-β(TGF-β)作为肾纤维化的靶点,它的激活使得肾小球的纤溶酶原激活物的活性显著降低,导致肾小球系膜基质沉积。研究[6]发现,TGF-β 与 PPARγ 之间也存在一定的联系,当RTEC 受到 TGF-β1攻击时,PPARγ 的表达和活性显著降低。我们前期研究[7]显示,调控PPARγ 基因的表达可减轻RTEC 的损伤程度,调控方式包括添加PPARγ 激动剂和抑制剂,以及通过转染慢病毒载体进行基因干扰[8],从而改变PPARγ 的表达水平。然而,在缺氧诱导的RTEC损伤中,有关PPARγ基因表达与细胞坏死性凋亡的关系,目前国内外尚未见报道。2019 年 10 月-2020 年 9 月,我们通过建立 RTEC坏死性凋亡模型,利用基因干扰技术沉默内源性和转染外源性 PPARγ 基因,观察了 PPARγ 基因过表达/沉默对缺氧诱导的RTEC 坏死性凋亡标志物表达的调控作用,现将结果报告如下。
1 材料与方法
1.1 细胞及培养方法 大鼠RTEC 细胞株(NRK-52E)购于上海细胞库,用含10%超滤胎牛血清和1%双抗的高糖DEME 培养基放置于37 ℃、50 mL/L CO2培养箱中培养。
1.2 细胞分组、转染及模型构建 将RTEC 细胞培养融合至80%左右时,将细胞培养瓶中的细胞消化为单个细胞,调整细胞密度传入细胞培养瓶并随机分为4组:对照组、缺氧损伤组、过表达组和沉默组。其中,过表达组细胞转染过表达PPAYγ 基因的外源性LV-PPARg 病毒,沉默组细胞转染沉默PPAYγ 基因的内源性LV-PPARg-RNAi 病毒。过表达组和沉默组的慢病毒载体均由上海吉凯基因公司构建,转染后经荧光显微镜和PCR 验证效果,转染效率达80%以上。待细胞再次生长融合至70%左右时,正常对照组不做任何处理,放置于培养箱中培养;缺氧损伤组、过表达组以及沉默组放置于含95%N2和5%CO2混合气体缺氧小室中,密封缺氧培养48 h。
1.3 各组细胞中PPARγ、RIP1、RIP3、TGF-β mRNA检测 采用实时荧光定量PCR 法。取各组细胞,TRIzol 法提取细胞总RNA,按照反转录试剂盒要求的标准条件反转录为cDNA。以cDNA 为模板,以GAPDH 为内参,应用荧光定量试剂盒进行荧光定量PCR。引物序列如下:GAPDH 上游引物为5′-GACATGCCGCCTGGAGAAAC-3′,下 游 引 物 为 5′-AGCCCAGGATGCCCTTTAGT-3′;PPARγ 上游引物为5′-GGAGCCTAAGTTTGAGTTTGCTGTG-3′,下游引物为 5′-TGCAGCAGGTTGTCTTGGATG-3′;RIP1上游引物为5′-CTTCTGCTCTCCTGGCTCATTGTG-3′,下游引物为5′-CGGTTGCTGATCTGGACGGTTG-3′;RIP3 上游引物为 5′-GGTGGTAGACAAGACCTCACTAATTCG-3′,下游引物为5′-ATGCGTCATTAACTCCTTCAGTCCTTC-3′;TGF-β 上游引物为 5′-ATGGTGGACCGCAACAACGC-3′,下游引物为 5′-CTGGCACTGCTTCCCGAATGTC-3′。建立 20.0 μL反应体系:2.5×Real MasterMix/SYBR solu-tion 9.0 μL,PCR Forward Primer(10 μmol/L)0.8 μL,PCR Reverse Primer(10 μmol/L)0.8 μL,cDNA 2 μL,超纯水7.4 μL。RT-qPCR 反应条件:50 ℃预热2 min,95 ℃激活10 min,95 ℃循环40次,每次15 s,60 ℃退火30 s。以2-ΔΔCt表示细胞中目的mRNA的相对表达量。
1.4 各组细胞中 PPARγ、RIP1、RIP3、TGF-β 蛋白检测 采用Western blotting 法。取出缺氧小室中的各组细胞,放入常规培养箱中复氧1 h,然后按照免疫沉淀测定(RIPA)蛋白裂解液说明书分别提取各组的总蛋白,以二喹琳甲酸(BCA)蛋白定量试剂盒测定蛋白浓度。将提取的各组蛋白,在4 ℃、12 000 r/min 离心 15 min 后分别吸取上清,按照 4:1 比例加入蛋白上样缓冲液轻柔吹打混匀,100 ℃水浴5 min变性。以GAPDH 为内参,每孔样本上样量为18 μg,行十二烷基硫酸钠—聚丙烯酰胺凝胶泳(SDS-PAGE),电泳结束后按照不同蛋白分子量确定转膜时间,采用湿转法转膜至聚偏二氟乙烯膜(PVDF),5%脱脂牛奶室温下封闭1.5 h 后,分别置于特异性一抗溶液中,4 ℃孵育16~18 h,后予以TBST溶液洗膜3次,每次10 min;再置于二抗溶液中室温下孵育1.5 h,TBST 溶液洗膜3次,每次10 min。以发光液覆盖PVDF 膜,扫描PVDF 显影,应用Alpha View 软件测定各蛋白的灰度值,以目的蛋白与内参蛋白灰度值的比值来反映各组目的蛋白的相对表达量。
1.5 统计学方法 采用SPSS22.0 统计软件。计量资料以表示,比较用单因素方差分析。P<0.05为差异有统计学意义。
2 结果
2.1 各组细胞中PPARγ、RIP1、RIP3、TGF-β mRNA相对表达量比较 各组细胞中PPARγ、RIP1、RIP3、TGF-β mRNA相对表达量比较见表1。
表1 各组细胞中PPARγ、RIP1、RIP3、TGF-β mRNA相对表达量比较()
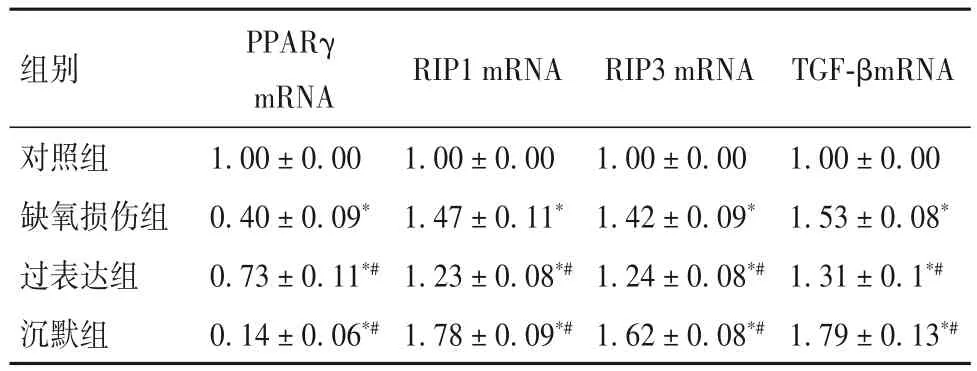
表1 各组细胞中PPARγ、RIP1、RIP3、TGF-β mRNA相对表达量比较()
注:与正常对照组相比,*P<0.05;与缺氧损伤组相比,#P<0.05。
组别RIP1 mRNA RIP3 mRNA TGF-βmRNA对照组缺氧损伤组过表达组沉默组1.00±0.00 1.53±0.08*1.31±0.1*#1.79±0.13*#PPARγ mRNA 1.00±0.00 0.40±0.09*0.73±0.11*#0.14±0.06*#1.00±0.00 1.47±0.11*1.23±0.08*#1.78±0.09*#1.00±0.00 1.42±0.09*1.24±0.08*#1.62±0.08*#
2.2 各组细胞中 PPARγ、RIP1、RIP3、TGF-β 蛋白相对表达量比较 各组细胞中PPARγ、RIP1、RIP3、TGF-β蛋白相对表达量比较见表2。
表2 各组细胞中PPARγ、RIP1、RIP3、TGF-β蛋白相对表达量比较()
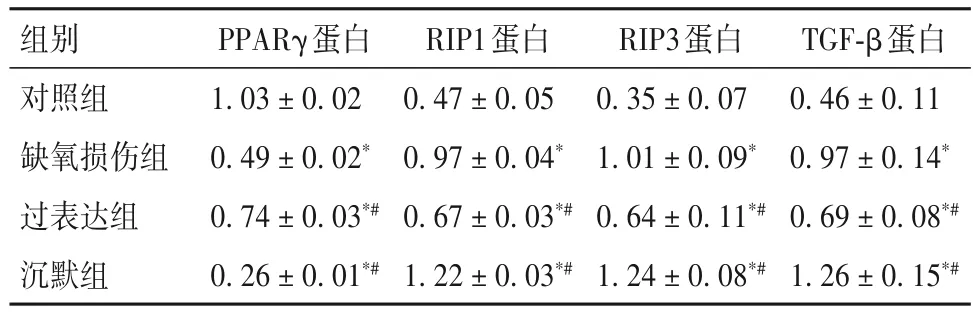
表2 各组细胞中PPARγ、RIP1、RIP3、TGF-β蛋白相对表达量比较()
注:与正常对照组相比,*P<0.05;与缺氧损伤组相比,#P<0.05。
组别对照组缺氧损伤组过表达组沉默组TGF-β蛋白0.46±0.11 0.97±0.14*0.69±0.08*#1.26±0.15*#PPARγ蛋白1.03±0.02 0.49±0.02*0.74±0.03*#0.26±0.01*#RIP1蛋白0.47±0.05 0.97±0.04*0.67±0.03*#1.22±0.03*#RIP3蛋白0.35±0.07 1.01±0.09*0.64±0.11*#1.24±0.08*#
3 讨论
RIF 是各类肾脏疾病进展至终末期肾病的必然途径和显著病理表现,其发生机制尚未完全明确,目前认为RTEC 损伤与RIF 的发生发展密切相关。研究[4]发现,在缺氧诱导的 RTEC 损伤中,激动剂罗格列酮可上调PPARγ 的蛋白表达而减轻RTEC 的损伤。在大鼠脓毒症模型中,激活PPARγ 可预防败血症大鼠的坏死和凋亡,保护心肌免受脓毒症所致损伤[9]。黄酮类非瑟酮通过调节 PPARγ 以减轻炎症和细胞凋亡,抑制基于MAPK 的分子机制从而保护心脏[10]。以上研究结果提示,PPARγ 的表达改变与坏死性凋亡关系密切。不仅如此,PPARγ 在调节肾脏生理功能中起重要作用[11],如PPARγ 基因突变可导致不正常的脂质和糖代谢、胰岛素抵抗,从而发展成肥胖引起的 2 型糖尿病[12~13];敲除 PPARγ 基因的小鼠出现肾功能不全,肌酐清除率降低,伴有纤维化和肾小管扩张[14];罗格列酮作为配体激活PPARγ,减轻了脓毒症时的炎症反应,减少肾脏细胞凋亡,改善了肾脏功能,对大鼠脓毒症急性肾损伤具有保护作用[15]。
凋亡和坏死是细胞死亡的两种病理类型。凋亡是程序控制的细胞死亡,在发育过程中和生理细胞更新过程中受到严格控制;坏死发生在创伤中,主要以不受控制的方式发生[16]。而坏死性凋亡是一种具有坏死特征的新型细胞死亡模式,在形态学上以细胞器肿胀和细胞膜破裂为特征,同时又是一种可调控的不依赖caspase 的非免疫细胞程序性细胞死亡,其中,RIP1、RIP3 是该途径的关键参与者[17~18]。RIPK3 的激活,对caspase-8 分子的抑制将外源性凋亡转变为细胞死亡的坏死模式;RIPK1 则抑制细胞凋亡和坏死的通过激酶独立的功能,对预防炎症有重要作用[19~20]。坏死性凋亡作为一种重要的细胞死亡模式,它的发生发展及阻断机制是一个复杂的过程,于肾脏而言,抑制肾脏细胞的坏死性凋亡可以减轻药物诱导的细胞毒性以及炎症反应的表达,对肾脏起到了一定的保护作用[21]。
上述研究提示,PPARγ 的表达水平和RTEC 坏死性凋亡均有可能参与了肾脏疾病的发生发展。PPARγ 作为参与调节肾脏许多重要生理功能的核转录因子,当肾脏受损时可能也会引起其表达水平的变化。而RIP1、RIP3作为坏死性凋亡的特异性蛋白,TGF-β 作为肾纤维化介导因子,它们与PPARγ之间是否存在联系?PPARγ 表达水平的改变是否会影响RTEC 坏死性凋亡?为了探讨这个问题,本研究通过建立缺氧诱导RTEC 坏死性凋亡模型,利用慢病毒沉默内源性和转染外源性PPARγ 基因,调控其在RTEC 中的表达水平,结果显示,与缺氧损伤组比较,过表达组细胞中PPARγ 的mRNA 及其蛋白表达量均显著增高,沉默组细胞中PPARγ 的mRNA及其蛋白表达量均显著降低;过表达组细胞中RIP1、RIP3、TGF-β的mRNA及其蛋白表达量均显著降低,而沉默组细胞中RIP1、RIP3、TGF-β 的mRNA及其蛋白表达量均显著增高,提示上调PPARγ 的基因表达可以减轻缺氧所致的RTEC 坏死性凋亡,而下调PPARγ的基因表达则加重RTEC坏死性凋亡。
综上所述,在缺氧诱导的RTEC 坏死性凋亡中,PPARγ基因表达降低;过表达PPARγ基因对缺氧诱导的RTEC 坏死性凋亡有抑制作用,而沉默PPARγ基因则有促进作用。
猜你喜欢
广东药科大学学报(2022年3期)2023-01-04
生物学通报(2022年1期)2022-11-22
中老年保健(2022年3期)2022-08-24
中老年保健(2022年3期)2022-08-24
中国种业(2021年11期)2021-11-25
现代临床医学(2021年6期)2021-11-20
天津医科大学学报(2021年2期)2021-03-29
天津医科大学学报(2021年2期)2021-03-29
昆明医科大学学报(2021年1期)2021-02-07
中国现代医药杂志(2020年10期)2020-12-14
