
复合型甘油激酶缺乏症一例临床资料及基因变异分析
2021-01-14 05:24崔清洋桑桂梅曹银利尚云唐成和
中国全科医学 2021年9期
崔清洋,桑桂梅,曹银利,尚云,唐成和
复合型甘油激酶缺乏症(complex glycerol kinase deficiency,CGKD)是一种呈X-连锁隐性遗传的相邻基因综合征,通常由Xp21染色体位点的部分缺失发展而来,包括导致甘油激酶缺乏症(glycerol kinase deficiency,GKD,OMIM# 307030)、先天性肾上腺发育不良(adrenal hypoplasia congenita,AHC,OMIM# 300200)、杜氏肌营养不良(Duchenne muscular dystrophy,DMD,OMIM# 310200)和智力残疾等多种发育缺陷的基因[1-3],常被误诊为其中的一种疾病。CGKD的症状与基因缺失的程度有关,可以在出生早期发病,男性发病占绝大多数。CGKD诊断主要依据临床和实验室检查结果,确诊主要依据基因检测发现Xp21位点缺失,并且可识别女性携带者。AHC症状较为严重,如不予以及时治疗,可在新生儿期死亡,而GKD症状一般为AHC掩盖;DMD早期可无明显的症状,仅在实验室检查结果中体现。本文回顾性分析了1例新生儿期CGKD患儿的临床资料和基因检测结果,以期加强临床医生对本病的认识。
1 病例简介
患儿,男,12 d,因呛水后哭闹、拒乳1 d,加重伴心律失常0.5 d而于2019-11-22入住新乡医学院第一附属医院。1 d前患儿呛水后出现哭闹不安,伴喉鸣及拒乳,无发热,无呕吐泡沫及呼吸急促,无四肢抖动及意识障碍,就诊于当地医院,诊断为“新生儿重症肺炎;新生儿高胆红素血症;心肌损害;新生儿低血糖?休克”,予以鼻导管吸氧,碳酸氢钠扩容纠酸、头孢哌酮舒巴坦及炎琥宁抗炎后喉鸣减轻;0.5 d前患儿出现心率加快(160次/min),伴血氧饱和度(SpO2)下降(具体数值不详)及皮肤发绀,予以经鼻持续气道正压通气(nCPAP)辅助通气后SpO2上升至90%以上,但40 min后心率下降至56次/min,且SpO2亦下降,予以胸外心脏按压、复苏囊加压辅助通气及肾上腺素和阿托品升高心率后,心率上升至90次/min,SpO2维持在85%左右,为进一步诊治由本院120接诊至本科。患儿系第二胎第二产,足月剖宫产,出生时无产伤史,Apgar评分不详。父母体健,非近亲结婚。患儿姐姐2岁,体健。入院体格检查:一般情况较差,反应较差,皮肤稍黄染,前囟平软,气管插管状态,口唇稍发绀,双肺呼吸音粗,可闻及痰鸣音,心率82次/min、律齐,心音稍低钝,腹部查体无异常,四肢肌张力及肌力可,原始反射未引出。外院实验室检查结果:肝功能:丙氨酸氨基转移酶(ALT)99 U/L(参考范围:9~50 U/L),天冬氨酸氨基转移酶(AST)603 U/L(参考范围:15~40 U/L),总胆红素(TBiL)179 μmol/L(参考范围:0~25 μmol/L),γ-谷氨酰转肽酶(γ-GT)1 125 μmol/L(参考范围:10~60 μmol/L),总胆汁酸(TBA)23 μmol/L(参考范围:0~10 μmol/L);心肌酶:乳酸脱氢酶(LDH)1 666 U/L(参考范围:313~618 U/L),肌酸激酶(CK)27 343 U/L(参考范围:55~170 U/L),肌酸激酶同工酶(CK-MB)1 400 U/L(参考范围:0~25 U/L)。入院后实验室检查:床旁心电图提示室性心动过速(呈快慢交替,心率快时血压下降,心率慢时血压正常);动脉血气分析:pH 7.51,氧分压(PO2)183 mm Hg(1 mm Hg=0.133 kPa)(参考范围:80~100 mm Hg),二氧化碳分压(PCO2)19.2 mm Hg(参考范围:35~45 mm Hg),K+7.6 mmol/L(参考范围:3.5~5.5 mmol/L),Na+131 mmol/L(参考范围:135~145 mmol/L),Ca2+0.85 mmol/L(参考范围:1.17~1.29 mmol/L),Cl-95 mmol/L(参考范围:99~107 mmol/L),血乳酸(Lac)3.1 mmol/L(参考范围:0.5~1.7 mmol/L),碱剩余(BE-)4.5 mmol/L(参考范围:-3.0~3.0 mmol/L),碳酸氢盐15 mmol/L(参考范围:22~26 mmol/L),提示过度通气及AG增高型代谢性酸中毒并呼吸性碱中毒;电解质:Na+122 mmol/L(参考范围:137~147 mmol/L),K+8.0 mmol/L(参考范围:3.5~5.3 mmol/L),Cl-89.2 mmol/L(参考范围:98~107 mmol/L),Ca2+2.11 mmol/L(参考范围:2.08~2.8 mmol/L),P 2.39 mmol/L(参考范围:0.81~1.15 mmol/L),Mg2+1.11 mmol/L(参考范围:0.7~1.0 mmol/L);肝功能:ALT 174 U/L,AST 780 U/L,TBiL 165.9 μmol/L,总蛋白(TP)70 g/L(参考范围:65~85 g/L),白蛋白(ALB)42 g/L(参考范围:40~55 g/L),γ-GT 1 014 μmol/L;心肌酶:LDH 9 840 U/L,CK 68 000 U/L,CK-MB 2 320 U/L;肌红蛋白(MYO)11 210 ng/ml(参考范围:0~121 ng/ml),肌钙蛋白I(cTnI)0.230 ng/ml(参考范围:0~0.034 ng/ml),B型脑钠肽前体(NT-proBNP)42 900 pg/ml(参考范围:0~250 pg/ml)。血常规、肾功能及凝血六项未见异常。胸片提示双肺纹理模糊;心脏彩超提示肺动脉压力41 mm Hg,左房室瓣及右房室瓣关闭不全(中度);复查心脏彩超提示左房室瓣关闭不全。入院诊断为:(1)新生儿肺炎;(2)新生儿阵发性室性心动过速;(3)心肌损害;(4)心功能不全;(5)新生儿电解质紊乱;(6)进行性肌营养不良。给予呼吸支持、抗感染、抗心律失常、保肝、营养心肌、纠正电解质紊乱及静脉营养治疗和调整呼吸机参数。针对心律失常的治疗:先给予阿托品0.01~0.03 mg/kg升高心率,但心率较快上升至242次/min,也较快下降至70~80次/min,再予以利多卡因复律,约1 h后仍未复律,给予葡萄糖酸钙对抗K+心肌毒性1 h后复律,心率维持在160次/min左右。因不能排除遗传代谢病可能,送检血及尿标本并行串联质谱及尿气相色谱-质谱检查,结果提示尿甘油明显升高,达309.5 μmol/L(参考范围:0~20.2 μmol/L),随机肾上腺皮质功能二项结果显示皮质醇2 μg/dl(参考范围:5~25 μg/dl),促肾上腺皮质激素(ACTH)258 pg/ml(参考范围:0~46 pg/ml),结合电解质和外院CK及本院CK检查结果,临床诊断为CGKD,因家庭经济困难患儿家属要求出院,也未予以激素口服治疗。
患儿出院1 d后因发热0.5 d再次入院,入院当天外送患儿及其父母和姐姐外周血标本至北京康旭医学检验所行基因检测。入院后给予抗感染治疗后发热缓解,继续补充3%氯化钠溶液纠正低钠血症,但收效甚微,仍未使用糖皮质激素及盐皮质激素。进一步检查:颅脑及双侧肾上腺MRI未见异常;皮质激素两项3次检查:8:00皮质醇22 μg/dl(参考范围:5~25 μg/dl),ACTH 124 pg/ml( 参 考 范 围:0~46 pg/ml);16:00皮质醇 26.8 μg/dl(参考范围:2.5~12.5 μg/dl),ACTH 93 pg/ml(参考范围:0~23 pg/ml);24:00皮质醇24 μg/dl(参考范围:1~8 μg/dl),ACTH 82.6 pg/ml;1 周后复查皮质激素两项3次检查:8:00皮质醇14 μg/dl,ACTH 64 pg/ml;16:00 皮 质 醇 20.3 μg/dl,ACTH 668 pg/ml,24:00皮质醇17 μg/dl,ACTH 141.0 pg/ml,均提示皮质醇未见明显异常,但ACTH不同程度升高。高血压四项(肾素、血管紧张素Ⅰ、血管紧张素Ⅱ和醛固酮)均未见异常。外送血标本经全外显子测序并通过CNV Seq验证发现受检者及其母亲Xp21.3-p21.1(chrX:25093883-34157184)区域存在缺失变异,长度约为9.063 Mb,缺失区域内基因包括MIR548F5、IL1RAPL1、FTHL17、MAGEB2、MAGEB3、GK、CXorf21、MAGEB5、VENTXP1、NR0B1、MAGEB1、PPP4R3CP、MAGEB4、MAGEB18、MIR4666B、DCAF8L1、FAM47A、MIR3915、MAGEB6、DCAF8L2、TAB3-AS1、TAB3、MAGEB10、DMD、MIR6134,未发现受检者父亲及其姐姐存在上述变异。上述缺失基因收录在OMIM数据库中的致病基因有IL1RAPL1、GK、NR0B1和DMD,且这4个基因全部整体缺失。至此,患儿CGKD诊断明确,因国内无氟氢可的松,出院后患儿口服醋酸氢化可的松(上海上药信谊药厂有限公司,生产批号:019190902),每次10~20 mg/m2,2次/d,后复查Na+较前稍升高(见表1)。
2 讨论
相邻基因综合征由相邻多个基因位点缺失所致,通常为偶发,可通过高分辨率核型分析或荧光原位杂交(FISH)诊断。相邻基因综合征相关的基因位于常染色体或性染色体上。常染色体相邻基因综合征(如22q11.2微缺失综合征和WAGR综合征-肾母细胞瘤、无虹膜和生长迟缓)通常有特征性临床表现,与特定代谢模式无关。CGKD是一种由Xp21染色体部分缺失引起的相邻基因综合征,目前尚无本病发病率的报道,但范瑞等[4]通过分析截至目前已报道的百例男性病例发现,其症状取决于缺失基因片段的大小[5]。AHC、DMD和GKD的基因位点距离最近,组合的形式常有AHC-GKDDMD、AHC-DMD、DMD-GKD,其中以复合型AHC-GKDDMD最为常见。
与常染色体相邻基因综合征不同,CGKD无特殊的畸形特征,患者多为男性,女性通常是无症状携带者,但女性可能有轻度至中度智力残疾,部分患者可能表现出CGKD的临床特征(如核型为45,X的Turner综合征患者)。
本例患儿入院前及入院时因明显的高肌酸激酶血症首先考虑DMD,且患儿出生时Na+、K+未见异常,入院时单纯高钾血症及低钠血症而无皮肤色素沉着不易与肾上腺皮质发育不良相关联,通过新生儿常规遗传代谢病筛查才得以确诊。
GKD的GK基因位于Xp21.3,长度约为98.1 kb,包括21个外显子,编码甘油激酶(糖异生途径的限速酶)。目前已发现的GK基因超过20种变异。AHC的NR0B1基因位于Xp21-Xp21.2,长度约为25.18 kb,包含2个外显子,编码孤儿核受体,存在于正在发育的肾上腺、性腺、下丘脑和垂体[6]。DMD基因是人类基因组中最大的基因之一,位于Xp21.2,长度约为2.26 Mb,包含79个外显子,60%的变异为大片段缺失,罕见部分重复、小缺失及单碱基替代变异[7]。IL1RAPL1基因位于Xp21.3-Xp22.1,长度约为1.39 kb,包含11个外显子,与DMD基因比邻,在神经系统高表达,与认知及精神发育有关[8]。
AHC是由位于X染色体上的DAX1基因(或NR0B1基因)缺失引起,可导致矿物质和糖皮质激素缺乏,但二者通常为CGKD的首发症状。AHC一般出现于出生后的最初几个月,常经过一段无症状时期,很少出现在儿童时期。AHC起病急,症状包括呕吐、发育不良、脱水、低血糖发作及由盐耗过多引起的休克。由于盐皮质激素缺乏症比其他激素缺乏症更为严重,出现时间也更早,因而耗盐症状通常是AHC的首要表现。
AHC患者的生化检查通常示脱水、低钠血症、高钾血症、酸中毒和低血糖,血清糖皮质激素、肾上腺皮质激素和雄激素水平极低,而血ACTH水平升高,或给予外源性ACTH后,血清皮质醇水平无升高。AHC患者可因垂体促肾上腺皮质激素分泌增加而出现色素沉着。本例患儿有脱水、休克、低钠血症及高钾血症,随机皮质醇下降,ACTH升高, 尽管颅脑及双侧肾上腺MRI无异常,无皮肤色素沉着表现,但基因结果证实NR0B1基因整体缺失,结合低钠及高钾血症,支持AHC的临床诊断。
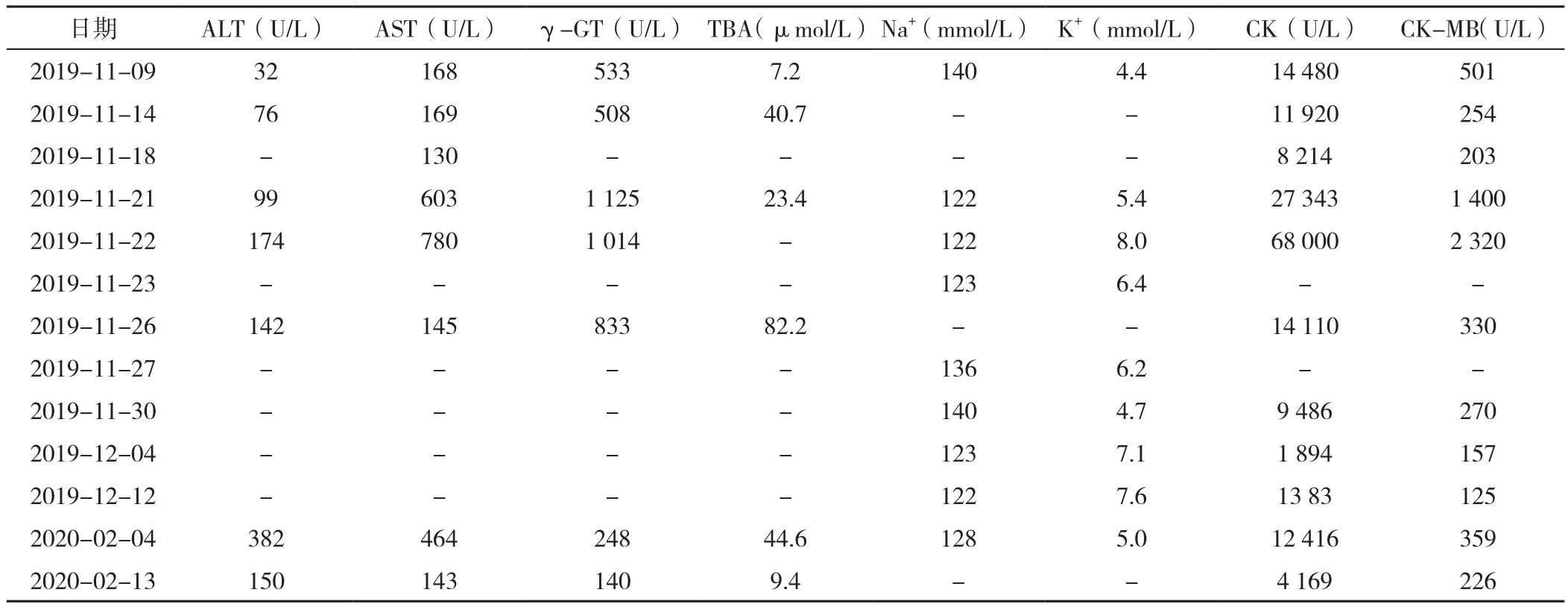
表1 患儿肝功能、电解质及心肌酶变化Table 1 Changes of liver function,electrolytes and myocardial enzymes in the neonatal patient
本病男婴早期表现为睾丸小或下丘脑和垂体混合起源的低促性腺素性功能减退引起的隐睾[9];血清黄体生成素、卵泡刺激素和睾酮水平极低,且单次促性腺激素释放激素注射反应低下,但其他垂体激素分泌正常;青春期前睾丸的组织学图像正常,青春期后睾丸结构异常,伴间质细胞增生,同时由于精子发生过程中可能出现异常而导致无精症[10]。罹患本病的男性青春期延迟,甚至可能不会出现第二性征,除非提供外源性激素供应,但其不育不能用任何方法治愈。本病女性携带者可能没有任何症状,但也可能存在青春期延迟。
甘油激酶催化甘油磷酸化反应生成磷酸甘油,在糖异生和游离脂肪酸再合成中是必需的。GKD患者在少年期或成年期由于能量平衡紊乱而发病。能量平衡紊乱的严重程度处于变化中,表现为从成年期无症状的高糖血症到儿童期危及生命的代谢危象[11]。GKD通常在年轻患者中症状更严重,但当与AHC相关时,肾上腺功能不全可能与GKD症状相似。GKD的症状最常发生在2~7岁,患儿可能出现呕吐、代谢性酸中毒、酮症性低糖血症、进行性嗜睡或意识不清等症状。低糖血症、酮症酸中毒和癫痫发作是GKD的三联征,而该病的发作通常出现在分解代谢情况下,如运动、进食过少、感染或其他严重疾病。由于大脑对葡萄糖的需求量与肝脏输出量成比例,因此儿童患者更容易出现低糖血症。
GKD最显著的表现是血及尿液中甘油明显升高[1,12]。甘油检测不是常规流程,普通实验室方法不能区分甘油和三酰甘油,只有血中三酰甘油水平非常高的患者才会检测。甘油酶活性可在白细胞、培养的皮肤成纤维细胞及肝、肾、小肠细胞和羊膜细胞等多种细胞中检测出。禁食、生酮饮食或运动试验等激发试验不推荐采用,因其不能阐明任何病因,且可能导致严重的并发症。突变携带者通常无症状,但其血中甘油水平可能略有升高,患者可能对禁食更为敏感。本例患儿在本院新生儿科住院期间无低糖血症,与文献报道不符,但尿中甘油水平明显升高,与文献报道一致[4]。
DMD属于退行性肌肉疾病,具有X-连锁隐性遗传特征,是儿童最常见的肌营养不良,病情呈进行性发展。DMD是由位于Xp21.2位点的编码抗萎缩蛋白的基因突变所致,可致进行性肌无力。DMD最初的症状发生在儿童早期,但部分症状如抬头落后可出现于婴儿期。DMD患儿开始步行的平均月龄为18个月,患病男孩表现出蹒跚的步态、脚趾走路和Gowers征,小腿肌肥大且可能有疼痛;到12岁时,患病男孩通常会丧失行走能力,需要轮椅才能移动。DMD所致肌无力涉及所有肌群,但眼外肌和远端肌功能通常不受影响,对于年龄较大的儿童,需要呼吸支持;同时肌功能不全会伴有挛缩和脊柱侧凸。
除了呼吸功能不全,另一个威胁DMD患者生命的因素是心血管问题,如扩张型心肌病和心律失常[13-14]。呼吸功能不全和心血管疾病是导致DMD患者在20岁或30岁死亡的主要原因。DMD患者也可能伴发智力迟钝,但程度通常较轻。DMD患者的血清 CK水平升高(15 000~3 5000 U/L)[15],在出生时也是如此,与肌肉退化有关。女性携带者通常没有DMD的临床症状,但80%的携带者血清CK水平升高。本例患儿转氨酶明显升高,为早期诊断肌营养不良的重要线索,临床遇到持续的转氨酶升高而不能用肝病解释时,应考虑到肌肉病的可能[16]。
相邻基因综合征是新发现的遗传病综合征,需要对每一个参与缺失的基因进行详细的分析,以了解每种综合征的症状机制。在基因测序方兴未艾的时代本病基因诊断并不困难。同时,临床医生需要了解该病的复杂特征,以便尽早做出正确及全面的诊断。
作者贡献:崔清洋进行文章的构思与设计,文章的可行性分析,文献/资料收集及整理,撰写论文;桑桂梅、曹银利、尚云进行论文的修订;唐成和负责文章的质量控制及审校,对文章整体负责,监督管理。
本文无利益冲突。
猜你喜欢
中国急救医学(2022年2期)2022-11-15
现代临床医学(2021年1期)2021-01-26
中华养生保健(2020年8期)2021-01-14
疯狂英语·新读写(2020年3期)2020-06-06
能源(2017年7期)2018-01-19
中学科技(2017年11期)2017-12-26
中国体育教练员(2017年2期)2017-07-31
临床医药文献杂志(电子版)(2017年11期)2017-05-17
广州化工(2016年8期)2016-09-02
中国民族医药杂志(2016年4期)2016-05-09
