
缺氧后适应中HSP90对补体介导的H9c2心肌细胞缺氧/复氧损伤的作用*
2020-12-03 13:46黄政涂荣会钟国强方存明马小林胡学俊
中国病理生理杂志 2020年11期
黄政, 涂荣会, 钟国强, 方存明, 马小林, 胡学俊
缺氧后适应中HSP90对补体介导的H9c2心肌细胞缺氧/复氧损伤的作用*
黄政1,2, 涂荣会2△, 钟国强2, 方存明1, 马小林1, 胡学俊1
(1宣城市人民医院心血管内科,安徽 宣城 242000;2广西医科大学第一附属医院心内科,广西 南宁 530022)
探讨缺氧后适应(HPC)中热休克蛋白90 (HSP90)对补体介导的大鼠H9c2心肌细胞缺氧/复氧(H/R)损伤的作用及机制。大鼠H9c2心肌细胞按处理方式不同分为7组:(1)对照组(常氧培养10 h);(2) H/R组(缺氧4 h后复氧6 h);(3) HPC组(缺氧4 h后行复氧5 min/缺氧5 min,重复3次,再复氧6 h);(4) HPC+格尔德霉素(GA)组(缺氧前20 min加入1 μmol/L的HSP90特异性抑制剂GA后行HPC);(5)阴性对照组(转染空载质粒后行HPC);(6) C3过表达组(转染C3a质粒后行HPC);(7) C5过表达组(转染C5a质粒后行HPC)。采用Hoechst 33242染色观察细胞形态变化,流式细胞术检测细胞凋亡, Western blot检测HSP90、C3a、C5a、NF-κB p65、TNF-α、IL-1β、IL-6、Bcl-2和Bax蛋白的表达。HPC在上调HSP90的同时显著减少H/R诱导的心肌细胞凋亡,并抑制C3a、C5a、NF-κB p65、TNF-α、IL-1β、IL-6和Bax的表达,增加Bcl-2的表达,上述效应可被GA阻断。过表达C3a和C5a后HPC对NF-κB p65表达及细胞凋亡的抑制作用被抵消。HPC中HSP90通过补体/NF-κB信号通路减轻H9c2心肌细胞H/R损伤。
缺氧后适应;缺血再灌注损伤;热休克蛋白90;补体;炎症
再灌注治疗是挽救缺血心肌,改善急性心肌梗死预后的最有效措施。然而,心肌血流恢复的同时也常伴随不同程度的缺血再灌注损伤,由其导致的心律失常、无复流、心肌顿抑、心肌细胞凋亡和坏死等极大地降低了再灌注治疗带来的获益。缺血后适应(hypoxic postconditioning, HPC)是被大量研究证实能够有效减轻心肌缺血再灌注损伤的方法,其机制可能与其调节多种促生存的信号转导通路及蛋白激酶有关[1-3]。近年来的研究表明,免疫炎症反应的过度激活是心肌缺血再灌注损伤的重要机制,缺血后适应可能通过抑制免疫炎症反应参与再灌注心肌的保护[4],然而具体机制仍有待阐明。
补体系统激活是心肌缺血再灌注损伤的重要特征。补体激活后除直接造成心肌细胞损伤外,还可激活NF-κB等信号转导通路,促进下游炎症细胞因子表达,诱导心肌细胞凋亡、坏死[5]。本课题组前期研究发现,缺血后适应能够上调热休克蛋白90 (heat shock protein 90, HSP90)表达,减轻缺血再灌注引起的心肌细胞凋亡、坏死。然而,HPC中HSP90对补体系统是否具有调节作用,目前尚不明确。
本研究通过建立离体心肌细胞缺氧/复氧(hypoxia/reoxygenation, H/R)损伤模型,实施IPC,体外模拟心肌缺血再灌注损伤,从细胞水平探讨缺血后适应心肌保护作用的机制。
材料和方法
1 材料
大鼠H9c2心肌细胞系购自中科院上海细胞库。DMEM培养基和胎牛血清购自Gibco; Lipofectamine 2000试剂盒购自Invitrogen; C3a和C5a过表达质粒载体购自Addgene;格尔德霉素(geldanamycin, GA)购自Sigma;大鼠抗肿瘤坏死因子α (tumor necrosis factor-α, TNF-α)、白细胞介素1β (interleukin-1β, IL-1β)、IL-6、C3a、C5a、NF-κB p65、Bcl-2、Bax和β-actin抗体购自北京博奥森; Hoechst 33242染色和BCA试剂盒购自上海碧云天; annexin V/APC细胞凋亡检测试剂盒购自江苏凯基。
2 方法
2.1细胞处理及分组H9c2心肌细胞在含10%胎牛血清的DMEM高糖培养基中,置于37℃、5% CO2、95%空气的培养箱中正常培养。细胞缺氧时更换为DMEM无糖培养基,并置于含5% CO2、95% N2混合气体的厌氧罐中。细胞复氧时再次更换为DMEM高糖培养基,置于37℃、5% CO2、95%空气的培养箱中。按处理方式不同分为7组:(1)对照(control)组(常氧培养10 h);(2) H/R组(缺氧4 h后复氧6 h);(3) HPC组(缺氧4 h后行复氧5 min/缺氧5 min,重复3次,再复氧6 h);(4) GA+HPC组(缺氧前20 min加入1 μmol/L GA后行HPC);(5)阴性对照(negative control, NC)组(转染空载质粒48 h后行HPC);(6) C3过表达组(C3组,转染C3a质粒载体后行HPC);(7) C5过表达组(C5组,转染C5a质粒载体后行HPC)。
2.2Hoechst 33242染色观察细胞形态取对数生长期的细胞接种于细胞爬片上,培养至约80%融合时进行分组处理。处理结束后弃去培养基, PBS漂洗后加入4%多聚甲醛,室温固定10 min,再加入终浓度为10 mg/L的Hoechst 33242染色液,避光染色5 min后加入抗荧光淬灭封片剂,荧光显微镜下观察细胞形态。
2.3流式细胞术检测细胞凋亡各组细胞胰酶消化,离心后收集细胞,用预冷的PBS漂洗2次,加入适量的1×结合缓冲液轻轻吹打重悬细胞,调整细胞浓度为1×109/L,取100 μL细胞悬液加入5 μL annexin V-APC,混匀,室温孵育15 min。再加入5 μL PI染色,流式细胞术检测细胞凋亡情况。
2.4Western blot检测蛋白表达各组细胞加入RIPA裂解液中冰上裂解30 min,14 000 r/min离心10 min后取上清液。采用BCA法测定蛋白浓度。取40 μg蛋白上样,SDS-PAGE分离后将蛋白转移至PVDF膜上,5%脱脂牛奶室温封闭2 h,加入Ⅰ抗溶液(浓度1∶1 000)中4℃孵育过夜,再加入Ⅱ抗溶液室温孵育1 h。经洗涤、显影、灰度扫描后,用Odyssey 3.0软件对蛋白条带进行灰度值测定。蛋白的表达量以目的条带与β-actin的灰度比值表示。
3 统计学处理
采用SPSS 20.0统计软件分析处理全部数据。计量资料以均数±标准差(mean±SD)表示;多组间比较采用单因素方差分析法,采用SNK-法进行组间两两比较。以<0.05为差异有统计学意义。
结果
1 HPC上调H9c2心肌细胞HSP90表达
与control组比较, H/R组HSP90表达显著增多(<0.05); HPC组HSP90表达较H/R组进一步增多(<0.05),而GA+HPC组HSP90表达较HPC组显著减少(<0.05),见图1。

Figure 1. Relative expression levels of HSP90 in different groups. Mean±SD. n=3. *P<0.05 vs control group; #P<0.05 vs H/R group; △P<0.05 vs HPC group.
2 HPC中HSP90对H9c2心肌细胞凋亡的影响
Hoechst 33342染色可见control组细胞核形态正常,呈圆形或椭圆形,淡蓝色,未见凋亡细胞; H/R组可见较多细胞核呈致密浓染,或呈碎块状致密浓染,颜色发白的凋亡细胞; HPC组凋亡细胞较H/R组明显减少; GA+HPC组凋亡细胞较HPC组明显增多,见图2。
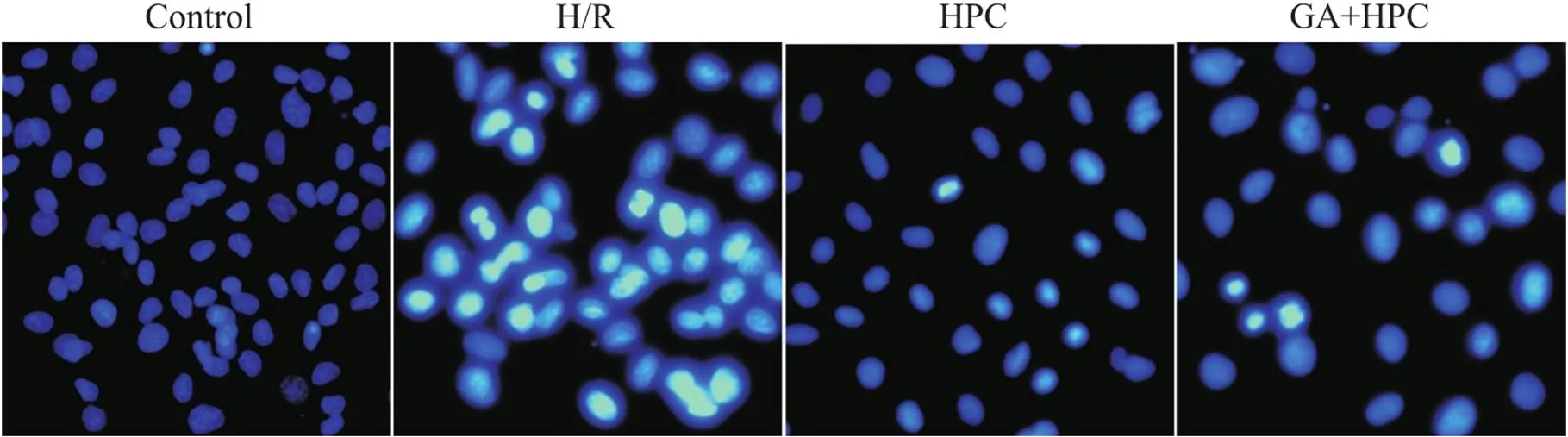
Figure 2. Morphological changes in the nucleus observed by Hoechst 33342 staining (×200). Apoptotic cells showed condensation of chromatin and nuclear fragmentations.
与Hoechst染色结果一致,流式细胞术检测H/R组的凋亡细胞均较control组显著增多(<0.05);与H/R组比较, HPC组的凋亡细胞显著减少(<0.05);而GA+HPC组的凋亡细胞较HPC组显著增多(<0.05),见图3。
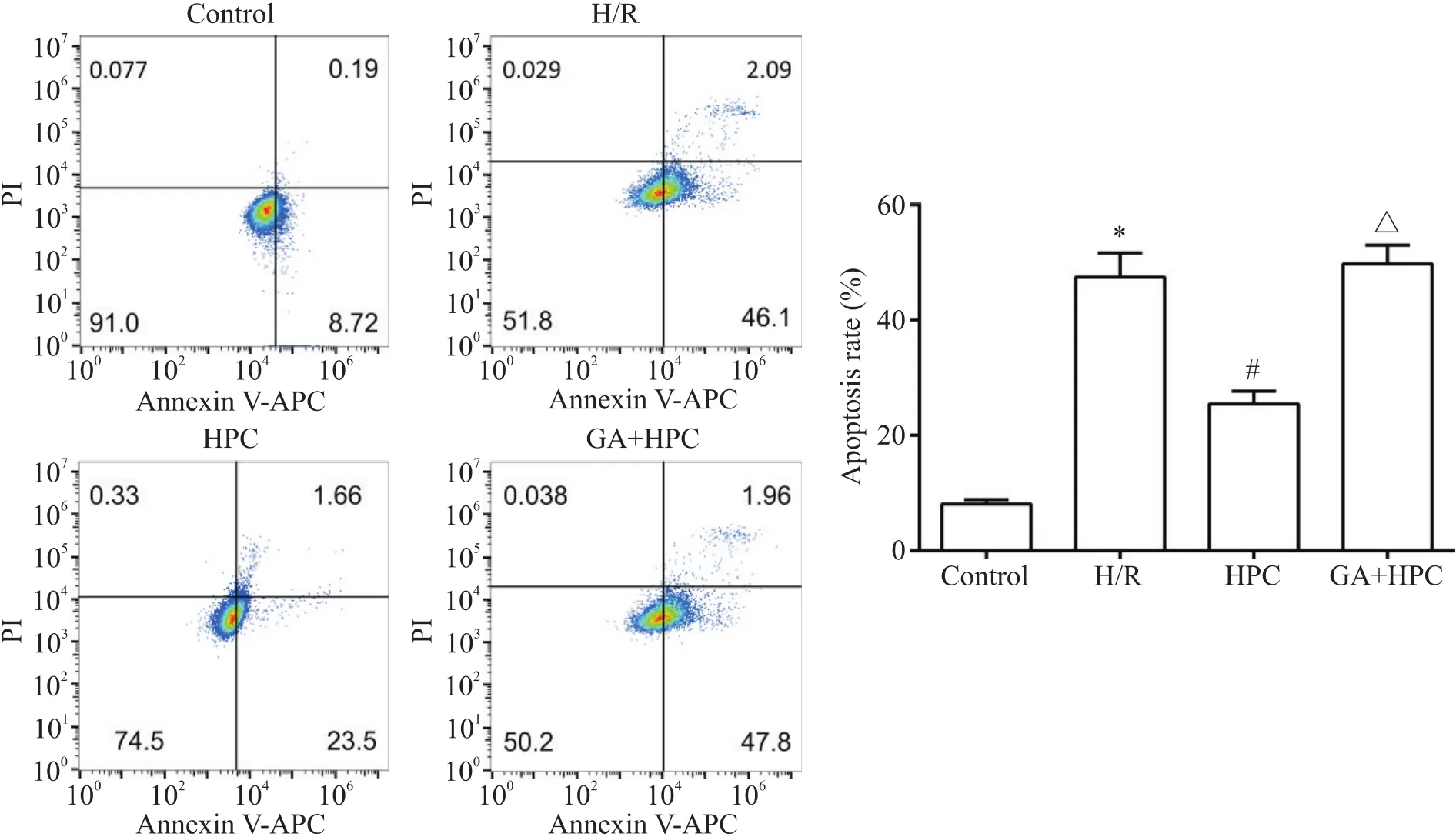
Figure 3. Apoptosis of the H9c2 cells determined by flow cytometry. Mean±SD. n=3. *P<0.05 vs control group; #P<0.05 vs H/R group; △P<0.05 vs HPC group.
3 HPC中HSP90对补体C3a/C5a和NF-κB表达的影响
H/R组补体C3a、C5a和NF-κB表达较control组显著增多(<0.05);与H/R组比较, HPC组C3a、C5a和NF-κB表达较H/R显著减少(<0.05);而GA+HPC组C3a、C5a和NF-κB表达较HPC组显著增多(<0.05),见图4。
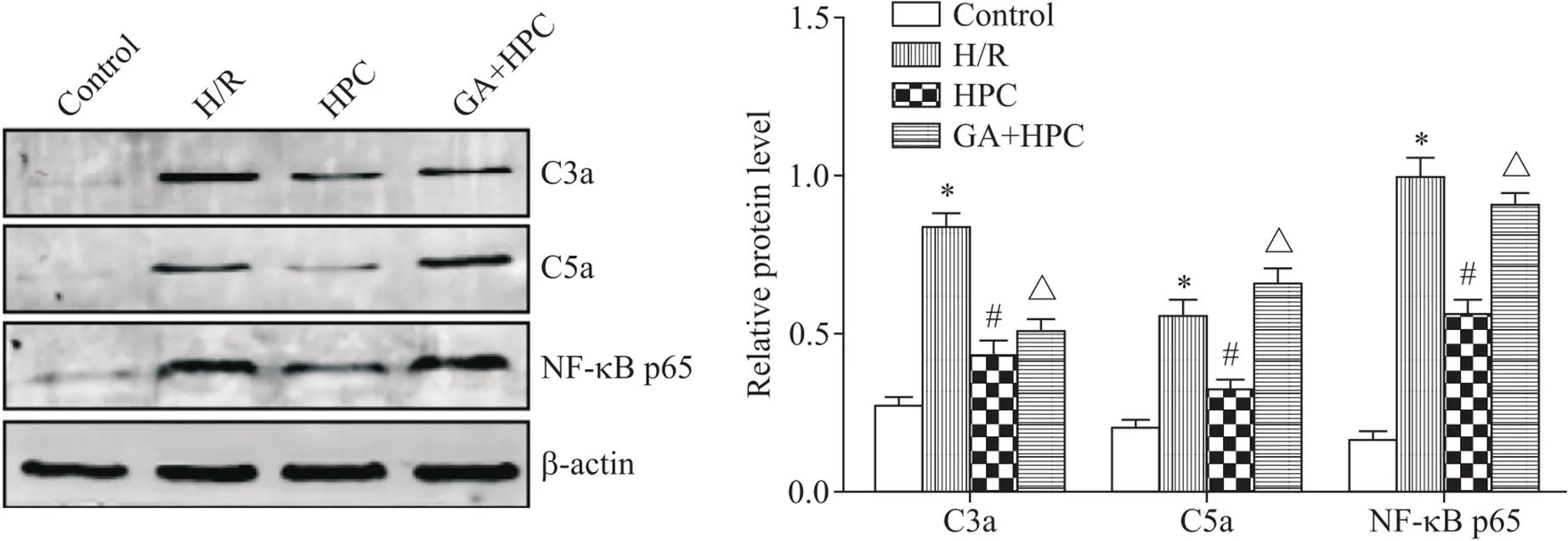
Figure 4. The protein expression of C3a, C5a and NF-κB was detected by Western blot. Mean±SD. n=4. *P<0.05 vs control group; #P<0.05 vs H/R group; △P<0.05 vs HPC group.
4 HPC中HSP90对炎症细胞因子表达的影响
与control组比较,H/R组TNF-α、IL-1β和IL-6表达显著升高(<0.05); HPC组TNF-α、IL-1β和IL-6表达显著低于H/R组(<0.05),而GA+HPC组的TNF-α、IL-1β和IL-6表达显著高于HPC组(<0.05),见图5。
5 HPC中HSP90对Bcl-2和Bax表达的影响
H/R组的Bcl-2表达显著低于control组(<0.05), Bax表达显著高于control组(<0.05);与H/R组比较, HPC组Bcl-2表达显著升高(<0.05), Bax表达显著下降(<0.05);而GA+HPC组Bcl-2表达较HPC组显著下降(<0.05), Bax表达较HPC组显著升高(<0.05),见图5。
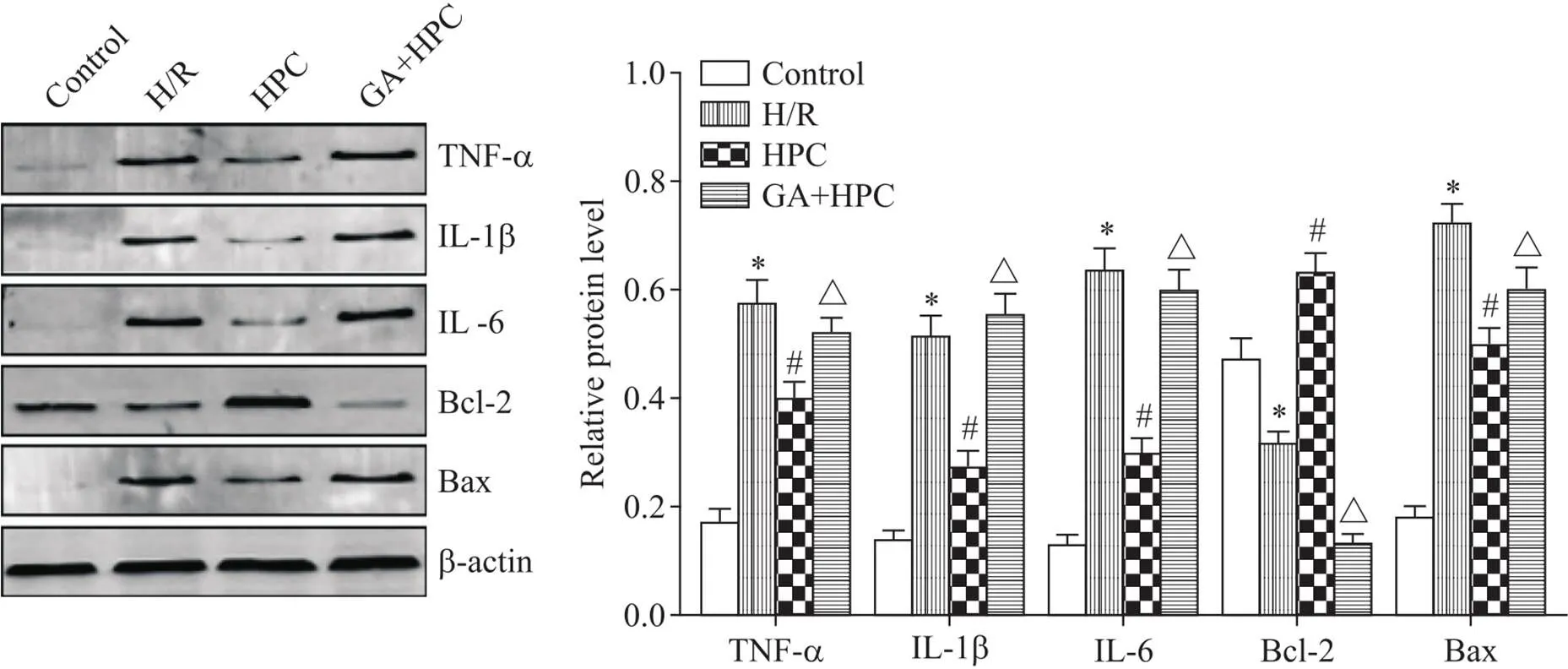
Figure 5. The results of Western blot showed the effects of HSP90 on the expression of TNF-α, IL-1β, IL-6, Bcl-2 and Bax in the H9c2 cells. Mean±SD. n=4. *P<0.05 vs control group; #P<0.05 vs H/R group; △P<0.05 vs HPC group.
6 HPC中补体-NF-κB信号通路对H9c2心肌细胞H/R损伤的影响
转染C3a过表达质粒载体后C3a的表达较阴性对照组显著增多(<0.05),同时NF-κB p65的表达显著增多(<0.05),心肌细胞凋亡率显著升高(<0.05)。转染C5a过表达质粒载体后C5a表达较阴性对照组显著增多(<0.05),同时NF-κB p65的表达显著增多(<0.05),H9c2心肌细胞的凋亡率显著升高(<0.05),见图6。
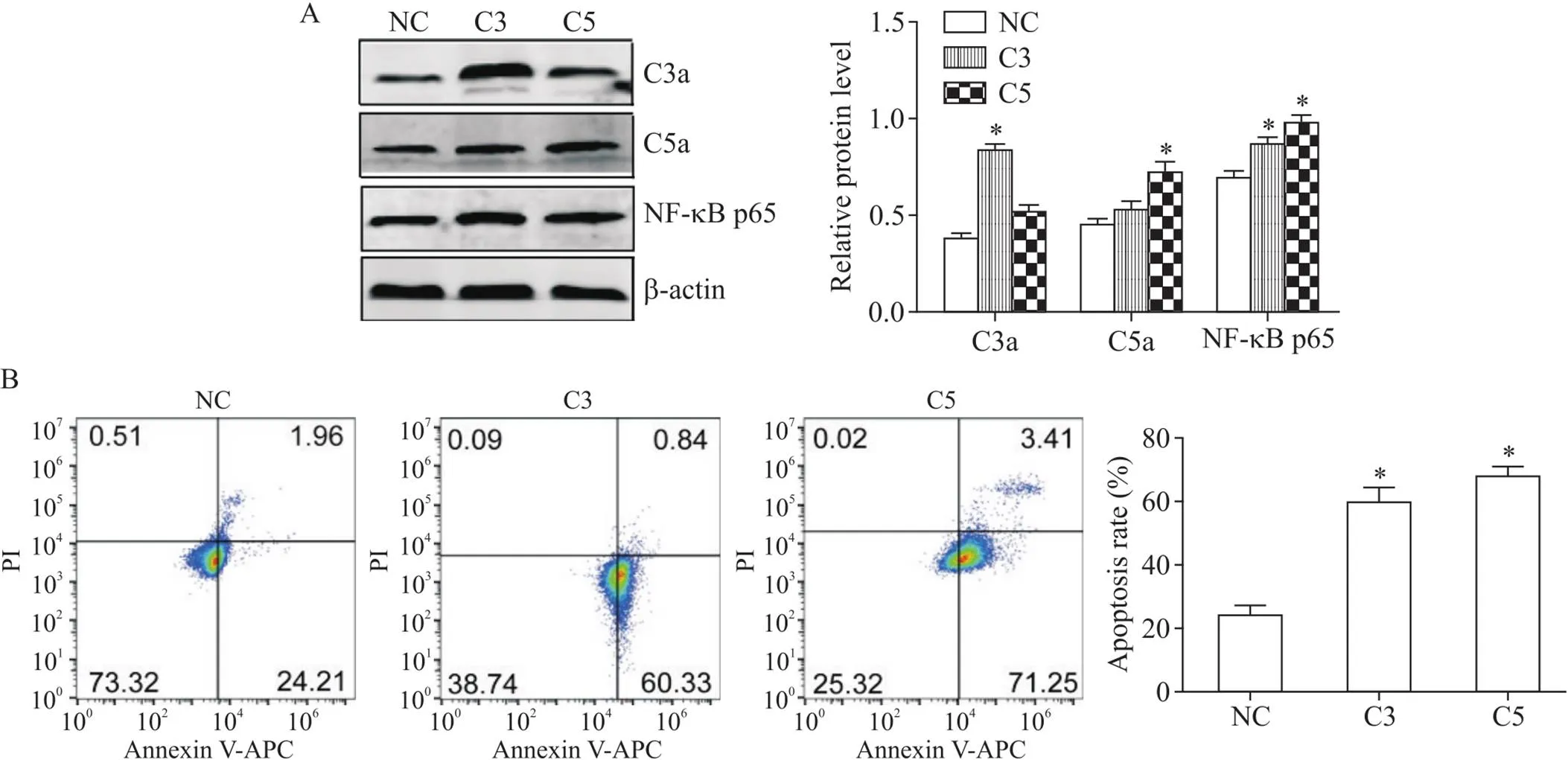
Figure 6. The effects of C3a and C5a over-expression on the expression of NF-κB in the H9c2 cells and H9c2 cell apoptosis. Mean±SD. n=3. *P<0.05 vs NC group.
讨论
本研究探讨了HPC减轻H9c2心肌细胞H/R损伤的作用及机制。研究发现, HPC在增加HSP90表达的同时显著减少H/R诱导的心肌细胞凋亡,并抑制补体系统激活及炎症细胞因子表达,而这种效应可被HSP90特异性抑制剂格尔德霉素阻断。研究结果提示,HPC可能通过上调HSP90表达减轻补体介导的H9c2心肌细胞H/R损伤及免疫炎症反应。
补体系统异常激活是缺血再灌注损伤的重要病理生理特征。补体激活后除通过攻膜复合体直接造成细胞损伤外,一些补体成分和裂解片段还可通过NF-κB、ERK1/2和JNK等信号转导通路促进TNF-α、IL-1和IL-6等炎症因子表达,间接导致靶细胞凋亡、坏死[5-6]。因此,抑制补体系统激活可能有助于减轻缺血再灌损伤、改善再灌注治疗的预后。已有多个研究报道,应用补体抑制剂或敲除补体基因可减少缺血再灌注动物模型心肌细胞凋亡、坏死,缩小心肌梗死面积,改善心脏功能[7-10]。Tanhehco等[11]的研究发现,缺血预处理可显著减少兔缺血再灌注局部心肌C1、C3、C8和C9的mRNA表达,减小心肌梗死面积。缺血预适应与缺血后适应均是被大量研究证实能够有效减轻心肌缺血再灌注损伤的措施,两者作用机制有许多类似之处。然而,缺血后适应能否作用于补体系统,目前尚无报道。本研究从离体细胞水平探讨HPC对心肌细胞补体系统的作用,研究发现HPC可显著减少H/R诱导的心肌细胞C3和C5a表达及细胞凋亡,提示抑制补体系统激活可能是HPC心肌细胞保护作用机制的一部分。
大量白细胞浸润引起过度放大的炎症级联反应是缺血再灌注损伤的主要机制之一。炎症反应过程中释放的一些炎症因子(如TNF-α、IL-1、IL-6等)可诱导靶细胞凋亡、坏死和血管内皮功能紊乱[12-14]。而补体系统对炎症反应具有促进作用。补体激活过程中释放的过敏毒素(如C3a、C5a等)和一些裂解片段具有很强的趋化效应,可吸引大量具有相应受体的白细胞向补体激活的区域聚集。另外,补体也可通过NF-κB信号转导通路,促进下游炎症细胞因子(如TNF-α、IL-1、IL-6等)表达,进一步增强炎症反应[15]。缺血后适应能够减少缺血再灌注心肌炎症细胞因子表达[4],但具体机制仍有待研究。本研究中我们观察到,HPC在抑制补体系统的同时伴随有NF-κB p65、TNF-α、IL-1β和IL-6表达及心肌细胞凋亡的显著减少,而过表达C3a和C5a后HPC对NF-κB和心肌细胞凋亡的抑制作用被抵消,提示HPC的心肌细胞保护效应与抑制补体-NF-κB信号通路介导的免疫炎症反应有关。
HSP90是细胞遭受不良刺激后新合成或合成增多的一种保护性蛋白,主要生物学功能是作为分子伴侣参与客户蛋白的正确折叠、复性和移位,从而维持细胞结构和功能稳定,防止细胞进一步损伤。Sreedhar等[16]的研究发现, HSP90抑制剂GA、根赤壳菌素和顺铂等可增强补体介导的Jurkat细胞裂解。我们的前期研究发现,缺氧(缺血)后适应通过上调 HSP90调节蛋白激酶C和线粒体缝隙连接蛋白43表达,减轻H/R心肌细胞及缺血再灌注心肌氧化应激损伤[17-18]。然而, HPC上调HSP90表达的同时是否对补体及其介导的免疫炎症反应产生影响,此前尚无报道。在本研究中,心肌细胞遭受H/R损伤刺激后HSP90表达增多,HPC进一步上调HSP90的同时显著抑制C3和C5a表达,并减少NF-κB、TNF-α、IL-1β、IL-6及促凋亡蛋白Bax的表达,增加抗凋亡蛋白Bcl-2的表达,而应用HSP90特异性抑制剂GA后这种效应被抵消,提示缺血后处理抑制补体激活、免疫炎症反应及细胞凋亡的机制可能与上调HSP90有关。事实上,缺血再灌注时激活补体和炎症反应的始动因素是细胞结构和功能的损伤,而后处理通过上调HSP90表达以及其他机制减轻了细胞损伤,进而减轻了补体激活和免疫炎症反应。
综上所述,HSP90介导HPC对H9c2心肌细胞的保护作用,其机制可能为抑制补体-NF-κB信号转导通路,减少补体-NF-κB信号通路介导的炎症细胞因子表达及细胞凋亡。
[1] Díaz-Ruíz JL, Macías-López A, Alcalá-Vargas F, et al. Redox signaling in ischemic postconditioning protection involves PKCε and Erk1/2 pathways and converges indirectly in Nrf2 activation[J]. Cell Signalling, 2019, 64:109417.
[2] Ren F, Mu N, Gao M, et al. Role of JNK signalling pathway and platelet-lymphocyte aggregates in myocardial ischemia-reperfusion injury and the cardioprotective effect of ischemic postconditioning in rats[J]. Mol Med Rep, 2018, 18(6):5237-5242.
[3]王茜,邓凤君,林焕冰,等. cAMP信号分子在缺血后适应心肌保护机制中的作用[J]. 中国病理生理杂志, 2011, 27(8):1496-1501.
Wang Q, Deng FJ, Lin HB, et al. Role of cAMP signaling in cardioprotective mechanism of ischemic postconditioning[J]. Chin J Pathophysiol, 2011, 27(8):1496-1501.
[4] Badalzadeh R, Baradaran B, Alihemmati A, et al. Troxerutin preconditioning and ischemic postconditioning modulate inflammatory response after myocardial ischemia/reperfusion injury in rat model[J]. Inflammation, 2017, 40(1):136-143.
[5] Chakraborti T, Mandal A, Mandal M, et al. Complement activation in heart diseases. Role of oxidants[J]. Cell Signalling, 2000, 12(9/10):607-617.
[6] Merle NS, Noe R, Halbwachs-Mecarelli L, et al. Complement system part II: role in immunity[J]. Front Immunol, 2015, 6:257.
[7] Busche MN, Stahl GL. Role of the complement components C5 and C3a in a mouse model of myocardial ischemia and reperfusion injury[J]. Ger Med Sci, 2010, 8:695-696.
[8] Chun N, Haddadin AS, Liu J, et al. Activation of complement factor B contributes to murine and human myocardial ischemia/reperfusion injury[J]. PLoS One, 2017, 12(6):e0179450.
[9] Riley RD, Sato H, Zhao ZQ, et al. Recombinant human complement C5a receptor antagonist reduces infarct size after surgical revascularization[J]. J Thoracic Cardiovasc Surg, 2000, 120(2):350-358.
[10] Yamamoto T, Tamaki K, Shirakawa K, et al. Cardiac Sirt1 mediates the cardioprotective effect of caloric restriction by suppressing local complement system activation after ischemia-reperfusion[J]. Am J Physiol Heart Circ Physiol, 2016, 310(8):H1003-H1014.
[11] Tanhehco EJ, Yasojima K, McGeer PL, et al. Preconditioning reduces myocardial complement gene expression[J]. Am J Physiol Heart Circ Physiol, 2000, 279(3):H1157-H1165.
[12] Gao C, Liu Y, Yu Q, et al. TNF-α antagonism ameliorates myocardial ischemia-reperfusion injury in mice by upregulating adiponectin[J]. Am J Physiol Heart Circ Physiol, 2015, 308(12):H1583-H1591.
[13] Zhu J, Huang J, Dai D, et al. Recombinant human interleukin-1 receptor antagonist treatment protects rats from myocardial ischemia-reperfusion injury[J]. Biomed Pharmacother, 2019, 111:1-5.
[14] Cheng C, Xu J M, YuT. Neutralizing IL-6 reduces heart injury by decreasing nerve growth factor precursor in the heart and hypothalamus during rat cardiopulmonary bypass[J]. Life Sci, 2017, 178:61-69.
[15] Ghosh S, Hayden M S. Celebrating 25 years of NF-κB research[J]. Immunol Rev, 2012, 246(1):5-13.
[16] Sreedhar AS, Nardai G, Csermely P. Enhancement of complement-induced cell lysis: a novel mechanism for the anticancer effects of Hsp90 inhibitors[J]. Immunol Lett, 2004, 92(1/2):157-161.
[17] Zhong GQ, Tu RH, Zeng ZY, et al. Novel functional role of heat shock protein 90 in protein kinase C-mediated ischemic postconditioning[J]. J Surg Res, 2014, 189(2):198-206.
[18] Tu RH, Li QJ, Huang Z, et al. Novel functional role of heat shock protein 90 in mitochondrial connexin 43-mediated hypoxic postconditioning[J]. Cell Physiol Biochem, 2017, 44(3):982-997.
Effects of HSP90 on complement-mediated hypoxia/reoxygenation injury of H9c2 cardiomyocytes during hypoxic postconditioning
HUANG Zheng1,2, TU Rong-hui2, ZHONG Guo-qiang2, FANG Cun-ming1, MA Xiao-lin1, HU Xue-jun1
(1,,242000,;2,,530022,)
To investigate the effects and mechanisms of heat shock protein 90 (HSP90) on complement-mediated hypoxia/reoxygenation (H/R) injury of rat H9c2 cardiomyocytes during hypoxic postconditioning (HPC).Rat H9c2 cardiomyocytes were divided into 7 groups according to different treatments:(1) control group (cultured for 10 h under normal oxygen);(2) H/R group (hypoxia for 4 h and reoxygenation for 6 h);(3) HPC group (3 cycles of 5 min H/R after hypoxia for 4 h, followed by reoxygenation for 6 h);(4) HPC+geldanamycin (GA) group (1 μmol/L HSP90 inhibitor GA was added 20 min before HPC);(5) negative control group (empty plasmid was transfected before HPC);(6) C3 over-expression group (C3a plasmid was transfected before HPC);(7) C5 over-expression group (C5a plasmid was transfected before HPC). Morphological changes of the H9c2 cells were detected by Hoechst 33242 staining. The effects of HPC on the apoptosis of H9c2 cells were examined by flow cytometry. The protein levels of HSP90, C3a, C5a, NF-κB p65, TNF-α, IL-1β, IL-6, Bcl-2 and Bax were determined by Western blot.With up-regulation of HSP90, HPC significantly reduced H/R-induced apoptosis of the H9c2 cells, inhibited the expression of C3a, C5a, NF-κB p65, TNF-α, IL-1β, IL-6 and Bax, and increased the expression of Bcl-2. These effects were blocked by GA. The inhibitory effects of HPC on NF-κB p65 expression and H9c2 cell apoptosis were offset after over-expression of C3a or C5a.HSP90 attenuates H/R injury of H9c2 cardiomyocytes by inhibiting complement-NF-κB signaling pathway during HPC.
Hypoxic postconditioning; Ischemia/reperfusion injury; Heat shock protein 90; Complement; Inflammation
R329.2+5; R363.2
A
10.3969/j.issn.1000-4718.2020.11.006
1000-4718(2020)11-1960-06
2020-02-11
2020-06-19
国家自然科学基金地区基金项目(No.81560068);宣城市科技计划项目(No.1817)
Tel: 13707885734; E-mail: 912881781@qq.com
(责任编辑:林白霜,罗森)
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
世界科学技术-中医药现代化(2022年2期)2022-05-25
世界科学技术-中医药现代化(2021年7期)2021-11-04
昆明医科大学学报(2021年8期)2021-08-13
江西农业学报(2021年4期)2021-04-20
昆明医科大学学报(2021年2期)2021-03-29
河南医学研究(2021年4期)2021-03-10
河南医学研究(2020年34期)2020-12-22
三农资讯半月报(2020年11期)2020-06-21
