
Aβ1-40通过激活MAPKs通路诱导血管平滑肌细胞发生炎症及表型转化*
2020-12-03 13:47张杰黄兴晓池菊芳
中国病理生理杂志 2020年11期
张杰, 黄兴晓, 池菊芳
Aβ1-40通过激活MAPKs通路诱导血管平滑肌细胞发生炎症及表型转化*
张杰, 黄兴晓▲, 池菊芳△
[绍兴市人民医院(浙江大学绍兴医院)心内科,浙江绍兴 312000]
探究β-淀粉样蛋白1-40 (Aβ1-40)对血管平滑肌细胞(VSMCs)的炎症反应、活力、迁移能力及表型转化的影响,并分析其机制。以不同浓度的Aβ1-40干预VSMCs适当时间。用CCK-8和Transwell实验评估细胞的活力及迁移能力;用Western blot检测细胞中白细胞介素1β (IL-1β)和肿瘤坏死因子α (TNF-α)等炎症因子水平, VSMCs表型转化标志蛋白α-平滑肌肌动蛋白(α-SMA)、骨桥蛋白(OPN)和Krüppel样因子4 (KLF4)的表达,以及丝裂原活化蛋白激酶(MAPKs)信号通路相关蛋白p-p38 MAPK、p-ERK1/2和p-JNK的蛋白水平。在Aβ1-40的作用下, VSMCs中炎症因子IL-1β和TNF-α水平显著升高,表型转化相关蛋白表达发生改变, α-SMA表达水平显著下降,而OPN和KLF4的表达水平显著升高(<0.05);且在一定浓度范围内, Aβ1-40可提高VSMCs的活力和迁移能力;另外,Aβ1-40处理可显著增加VSMCs中p-p38 MAPK、p-ERK1/2和p-JNK的蛋白水平(<0.05)。应用MAPKs相应的抑制剂可显著降低IL-1β和TNF-α表达水平(<0.05),并抑制细胞表型转化,即α-SMA表达水平显著升高, OPN和KLF4表达水平显著下降(<0.05)。Aβ1-40可通过激活MAPKs通路诱导VSMCs发生炎症反应和表型转化。
β-淀粉样蛋白1-40;血管平滑肌细胞;炎症;表型转化;MAPKs信号通路
β-淀粉样蛋白(amyloid β-protein, Aβ)在脑内的沉积是阿尔茨海默病(Alzheimer disease, AD)的重要发生机制之一,其中Aβ1-40和Aβ1-42在AD的病理过程中显得尤为重要[1-2]。且随着研究的深入,上述两种淀粉样蛋白在AD中的具体定位日渐清晰。Aβ1-40主要通过与脑血管较高的亲和力而沉积于血管,造成大脑淀粉样血管病(cerebral amyloid angiopathy, CAA),这是一种AD的标志性病变,进而促进AD的进展[3]; Aβ1-42则更多的是沉积在脑组织而发挥其作用[4]。然而Aβ1-40与脑血管的亲和力并无特异性,预示其可能不仅仅只在脑血管中沉积,还可能在外周血管中也发挥促炎等作用。一项关于Aβ在全身血浆及组织中水平的研究显示,在动脉粥样硬化患者的主动脉壁上,Aβ1-40和Aβ1-42分别以75.3 ng/g和0.7 ng/g的含量存在于动脉粥样硬化斑块之中[5]。同时,最新资料也表明AD与心血管疾病(cardiovascular diseases, CVD)存在许多共享的病理机制, Aβ1-40在CVD中也同样发挥着重要作用,而针对Aβ1-40代谢可能将成为一种有效的CVD治疗方式[6]。但目前为止,仅有少量关于Aβ1-40与心血管疾病关系的研究,而细分到动脉粥样硬化的研究更是寥寥无几。据此,本研究主要探讨Aβ1-40与动脉粥样硬化的关系。另外,由于血管平滑肌细胞(vascular smooth muscle cells, VSMCs)在动脉粥样硬化中发挥着十分关键的作用,因此本研究观察Aβ1-40对VSMCs炎症和表型转化的影响,并探讨丝裂原活化蛋白激酶(mitogen-activated protein kinases, MAPKs)信号通路[包括细胞外信号调节激酶(extracellular signal-egulated kinase, ERK)、c-Jun氨基末端激酶(c-Jun NH2-terminal kinase, JNK)和p38 MAPK]在其中的作用。
材料和方法
1 实验材料
VSMCs购自中国科学院。DMEM培养基购自Sigma;胎牛血清购自Gibco; Aβ1-40购自Abcam; SB203580、U0126、SP600125和CCK-8试剂盒均购自Mce; Transwell小室购自Corning; BCA试剂盒和ECL发光液购自碧云天公司;抗白细胞介素1β (interleukin-1β,IL-1β)、肿瘤坏死因子α (tumor necrosis factor-α, TNF-α)、α-平滑肌肌动蛋白(α-smoth muscle actin, α-SMA)、骨桥蛋白(osteopontin, OPN)、Krüppel样因子4 (Krüppel-like factor 4, KLF4)、p38 MAPK、p-p38 MAPK、ERK1/2、p-ERK1/2、JNK和p-JNK抗体均购自Abcam。
2 实验方法
2.1细胞培养用含10%胎牛血清和1%青-链霉素的DMEM高糖培养基(Sigma)培养VSMCs。细胞置于37℃、5% CO2加湿培养箱之中。
2.2细胞干预为探究不同浓度Aβ1-40对VSMCs的作用,将VSMCs随机分为4组:(1)对照(control)组,给予正常培养基;(2) 0.5 µmol/L Aβ1-40组,培养基中含0.5 µmol/L Aβ1-40;(3) 2.5 µmol/L Aβ1-40组,培养基中含2.5 µmol/L Aβ1-40;(4) 5.0 µmol/L Aβ1-40组,培养基中含5.0 µmol/L Aβ1-40。为探究MAPKs信号通路在Aβ1-40所致VSMCs炎症和表型转化中的作用,将VSMCs随机分为5组:(1)对照组,给予正常培养基;(2) Aβ1-40组,培养基中含2.5 µmol/L Aβ1-40;(3) Aβ1-40+SB203580组,在用2.5 µmol/L Aβ1-40干预VSMCs之前,用20 µmol/L p38 MAPK抑制剂SB203580预处理细胞30 min;(4) Aβ1-40+U0126组,在用2.5 µmol/L Aβ1-40干预VSMCs之前,用20 µmol/L ERK1/2抑制剂U0126预处理细胞30 min;(5) Aβ1-40+SP600125组,在用2.5 µmol/L Aβ1-40干预VSMCs之前,用20 µmol/L JNK抑制剂SP600125组预处理细胞30 min。干预适当时间后,终止培养,进行后续实验分析。
2.3CCK-8实验检测细胞活力将VSMCs以每孔6 000个的密度种植于96孔板中,待细胞贴壁之后,分别予以不同浓度的Aβ1-40进行干预。0、24、36和48 h之后,每孔避光加入10 μL的CCK-8液,继续在培养箱中培养2 h,最后用酶标仪在450 nm波长下测定吸光度()值。
2.4Transwell实验检测细胞的迁移能力将Transwell小室置于24孔板中,上室以5×104细胞密度种植VSMCs(无血清培养基),下室加入600 μL含5%胎牛血清及不同浓度Aβ1-40的培养基,培养24 h后进行结晶紫染色,随后置于倒置显微镜下观察并计数。
2.5Western blot检测蛋白水平用RIPA裂解液提取各实验组蛋白,随后用BCA试剂盒进行蛋白浓度测定并配平。以10% SDS-PAGE进行蛋白分离,之后转膜。用5%脱脂奶粉室温封闭PVDF膜1 h,随后加入相应I抗(抗IL-1β、TNF-α、α-SMA、OPN、KLF4、p38 MAPK、p-p38 MAPK、ERK1/2、p-ERK1/2、JNK和p-JNK抗体)4℃孵育过夜。第2天加入相应II抗室温孵育2 h,最后加ECL发光液于自动曝光机下进行曝光。
3 统计学处理
用SPSS 26.0和GraphPad Prism 8软件进行统计分析。实验数据以均数±标准差(mean±SD)表示,组间均数比较采用单因素方差分析及Tukey法。以<0.05为差异有统计学意义。
结果
1 Aβ1-40可促进VSMCs发生炎症反应
以不同浓度的Aβ1-40干预血管平滑肌细胞24 h之后,评估各组细胞炎症水平。结果显示,与对照组相比,随着Aβ1-40浓度的逐渐增加,VSMCs的炎症因子(IL-1β和TNF-α)水平逐渐升高(<0.05),见图1。这提示Aβ1-40可促进VSMCs发生炎症反应,且呈浓度依赖性。
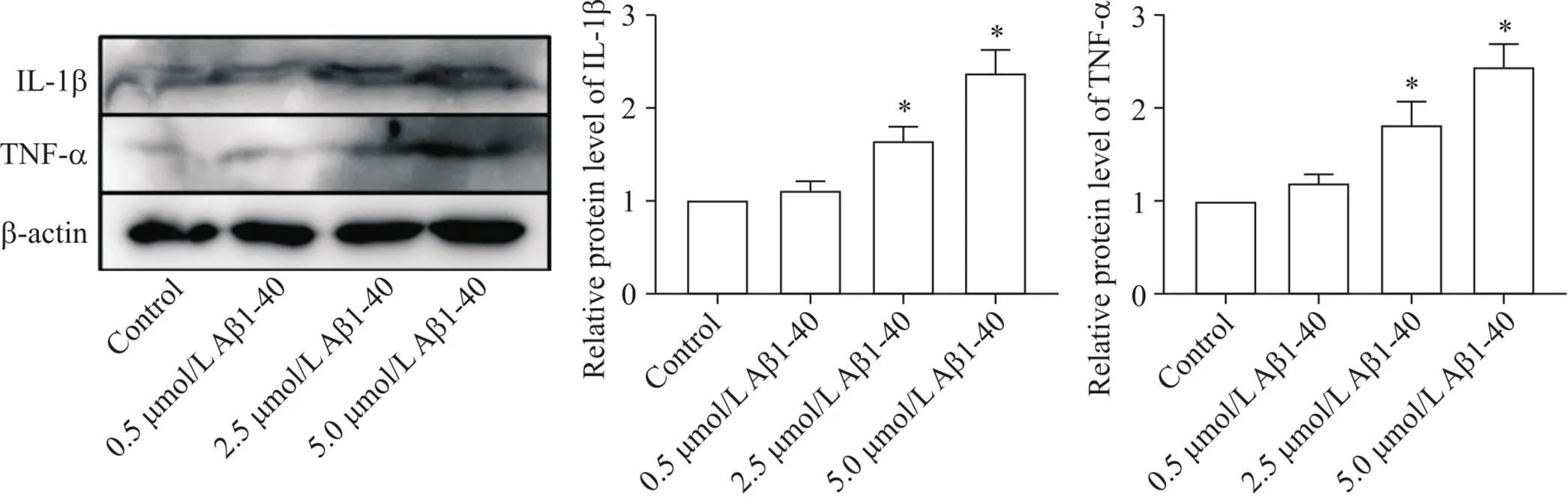
Figure 1. Effects of Aβ1-40 on inflammation of VSMCs. The protein expression levels of IL-1β and TNF-α were detected by Western blot. Mean±SD. n=3. *P<0.05 vs control group.
2 Aβ1-40对VSMCs活力及迁移能力的影响
以不同浓度的Aβ1-40干预血管平滑肌细胞,分别于0 h、24 h、36 h和48 h之后用CCK-8实验检测细胞活力。结果显示,在一定浓度范围内(<2.5 µmol/L),随着Aβ1-40浓度的增加,VSMCs的活力逐渐升高(<0.05);但当以更高浓度(5 µmol/L)的Aβ1-40处理VSMCs时, VSMCs活力下降,见图2A。用不同浓度Aβ1-40干预VSMCs 24 h后, Transwell实验结果显示,一定浓度范围内(<2.5 µmol/L),随着Aβ1-40浓度的增加,VSMCs的迁移能力逐渐增强(<0.05);而以更高浓度(5 µmol/L)的Aβ1-40干预细胞,VSMCs的迁移能力下降,见图2B。这些结果提示,在一定浓度范围内,Aβ1-40可增强VSMCs的活力和迁移能力。
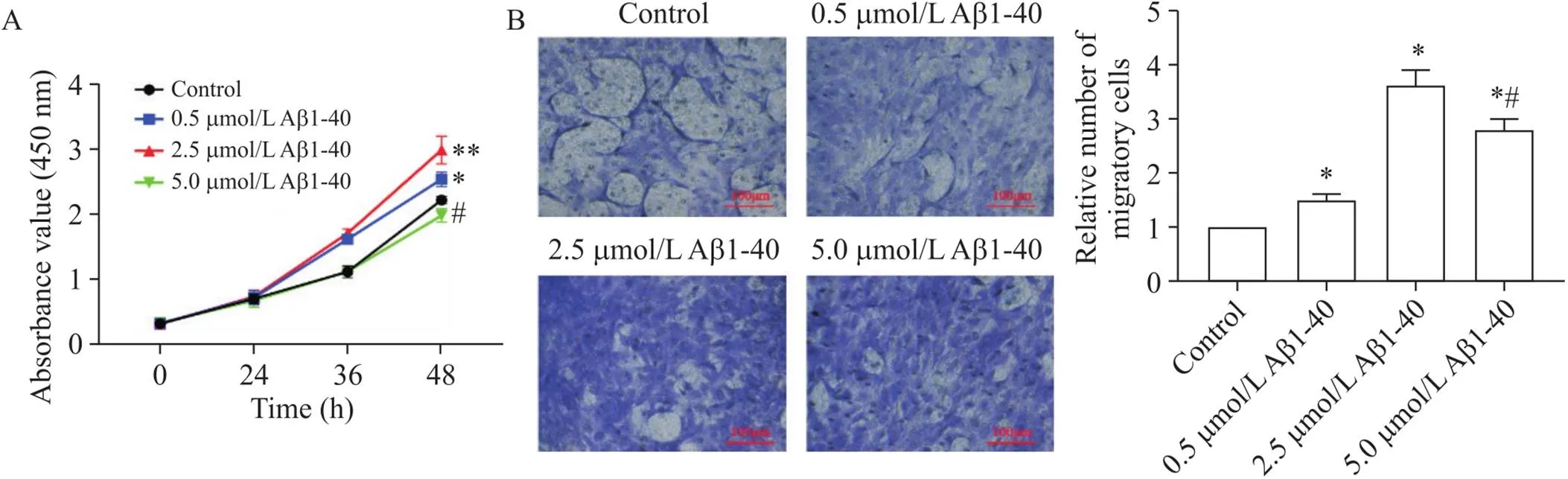
Figure 2. Effect of Aβ1-40 on the viability and migration ability of VSMCs. A: the viability of VSMCs treated with different concentrations (0.5, 2.5 and 5.0 µmol/L) of Aβ1-40 was measured by CCK-8 assay; B: the migration ability of VSMCs was detected by Transwell assay. Mean±SD. n=3. *P<0.05, **P<0.01 vs control group; #P<0.05 vs 2.5 µmol/L Aβ1-40 group.
3 Aβ1-40可促进VSMCs发生表型转化
以不同浓度的Aβ1-40干预VSMCs 24 h之后,检测各组细胞表型转化指标α-SMA、OPN和KLF4的表达水平。结果显示,与对照组相比,随着Aβ1-40浓度的逐渐增加, α-SMA表达水平逐渐下降,而OPN和KLF4的表达水平逐渐升高(<0.05),见图3。这提示Aβ1-40可促进VSMCs发生表型转化,且呈浓度依赖性。
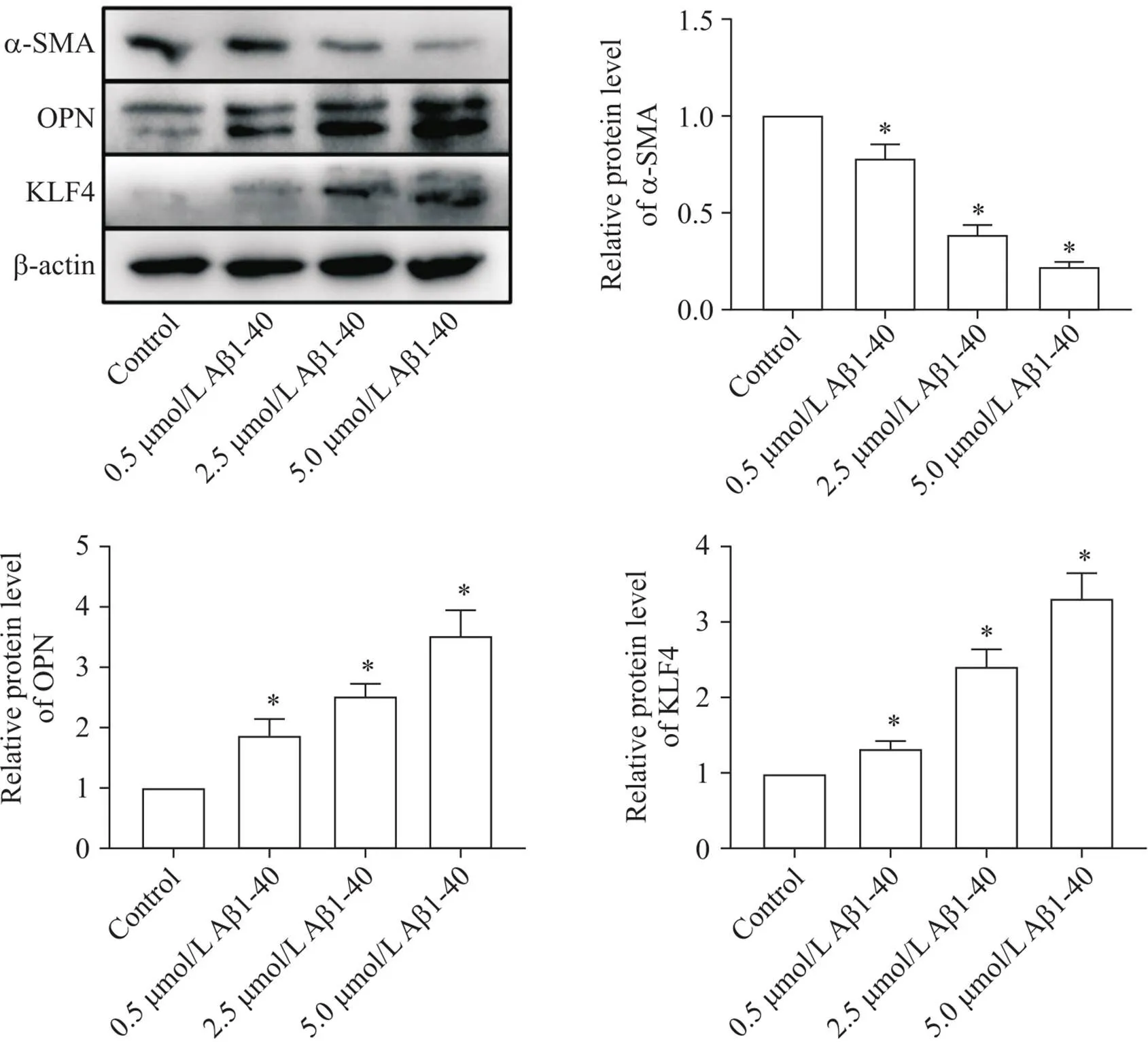
Figure 3. Effect of Aβ1-40 on phenotypic switching of VSMCs. The protein expression levels of α-SMA, OPN and KLF4 were determined by Western blot. Mean±SD. n=3. *P<0.05 vs control group.
4 Aβ1-40通过MAPKs信号通路发挥其作用
以不同浓度的Aβ1-40干预VSMCs 24 h之后,检测各组细胞MAPKs信号通路活性。Western blot结果显示,随着Aβ1-40浓度的逐渐提高, p-p38 MAPK、p-ERK1/2和p-JNK蛋白水平均逐渐增加(<0.05),而总p38 MAPK、ERK1/2和JNK的蛋白水平无显著变化,提示p38 MAPK、ERK1/2和JNK通路的逐渐激活,见图4。该结果表明MAPKs系列通路参与了Aβ1-40对VSMCs的作用过程。

Figure 4. Effect of Aβ1-40 on activation of MAPKs pathway in VSMCs. The relevant protein levels of p38 MAPK, p-p38 MAPK, ERK1/2, p-ERK1/2, JNK and p-JNK were detected by Western blot. Mean±SD. n=3. *P<0.05 vs control group.
5 抑制MAPKs信号通路可缓解Aβ1-40诱导的VSMCs炎症反应和表型转化
为进一步验证MAPKs信号通路在Aβ1-40诱导的VSMCs炎症和表型转化中的作用,我们使用p38 MAPK、ERK1/2和JNK的特异性抑制剂SB203580、U0126和SP600125对细胞进行预处理,之后再以2.5 µmol/L Aβ1-40干预VSMCs。结果显示,与Aβ1-40组相比,3种通路抑制剂均能显著降低VSMCs的炎症水平(IL-1β和TNF-α水平下降),并抑制细胞的表型转化(α-SMA表达升高,OPN和KLF4表达下降),见图5。此结果进一步证实了MAPKs信号通路在Aβ1-40诱导的VSMCs的炎症和表型转化中的作用,并且阻断上述通路可显著抑制VSMCs的功能变化。
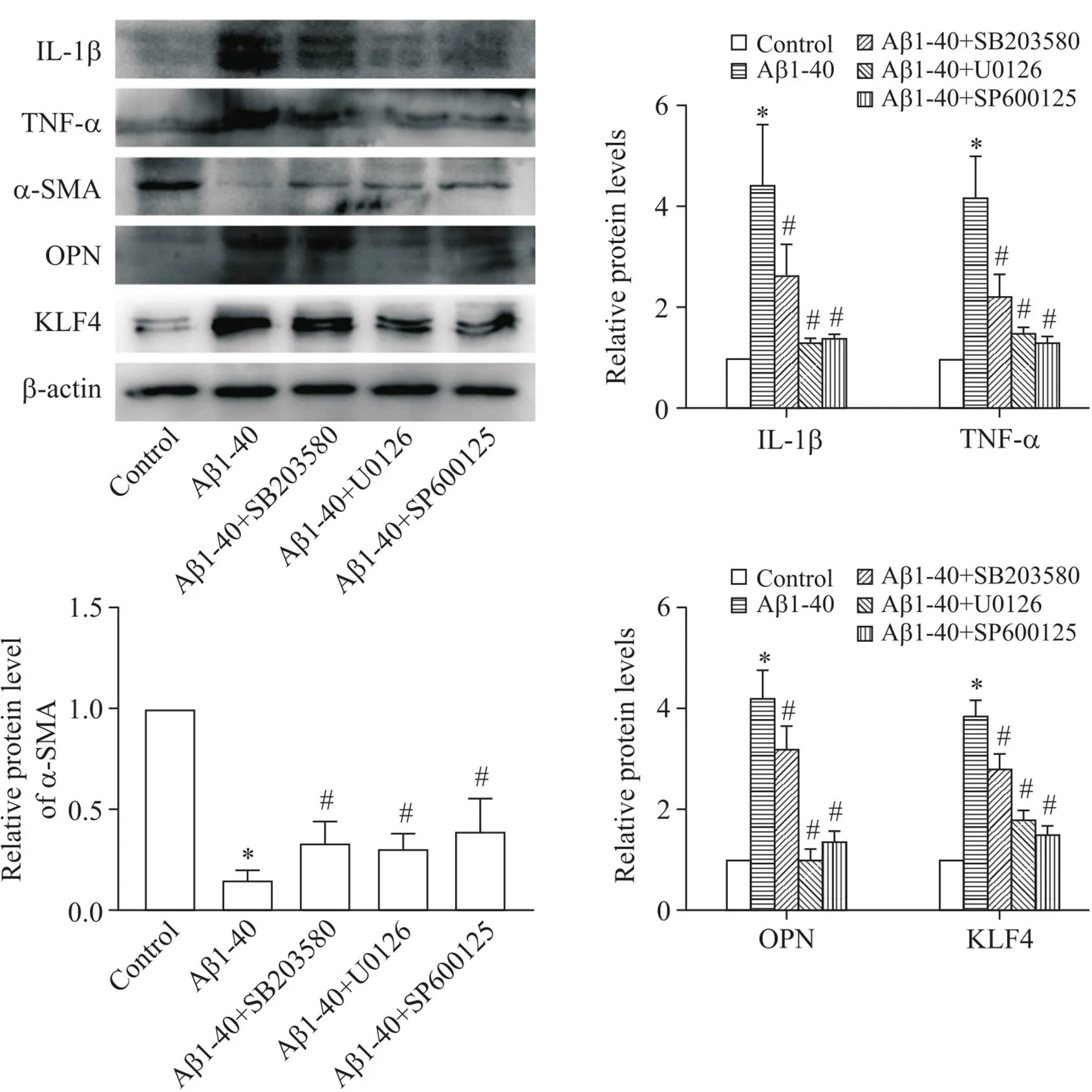
Figure 5. Effects of specific inhibitors of MAPKs signaling pathway on Aβ1-40-induced changes in VSMCs. The protein levels of IL-1β, TNF-α, α-SMA, OPN and KLF4 were determined by Western blot. Mean±SD. n=3. *P<0.05 vs control group; #P<0.05 vs Aβ1-40 group.
讨论
动脉粥样硬化是一种常见的心血管疾病,其发病机制复杂多样,但可以明确的是炎症和VSMCs的增殖、迁移及表型转化在其中发挥着重要作用[7]。而同为系统退行性病变, AD被越来越多的研究证明与动脉粥样硬化之间存在许多共性及联系[8]。Aβ1-40作为AD的标志性蛋白之一,因其对冠心病和心力衰竭等心血管疾病的促进作用,吸引了大量心血管学者的关注[9-10],但目前尚无明确研究揭示其在动脉粥样硬化中的作用。本实验用不同浓度Aβ1-40处理VSMCs,结果发现Aβ1-40可以导致VSMCs发生炎症反应,促进VSMCs增殖和迁移并诱导其发生表型转化,且存在浓度依赖性。
MAPKs是一类广泛表达于真核生物的丝/苏氨酸蛋白激酶,主要包括ERK1/2、JNK和p38 MAPK三个家族成员[11]。MAPKs可接受多种细胞外信号刺激,并通过逐层级联磷酸化激活导致下游一系列反应,进而调控细胞炎症、增殖和分化等活动[12]。我们通过对MAPKs家族磷酸化水平的检测及相应抑制剂的应用发现, ERK1/2、JNK及p38 MAPK信号通路均参与了Aβ1-40诱导的VSMCs变化,而抑制相应的通路则可降低VSMCs的炎症水平和表型转化程度。
综上所述,本研究证明了Aβ1-40可诱导VSMCs发生炎症反应和表型转化,且在一定浓度内可促进VSMCs的活力和迁移,而这个过程中MAPKs信号通路发挥了重要作用。我们的结果可能会为动脉粥样硬化的治疗提供一种新思路。然而,动物及临床等进一步研究还有待开展。
[1] Siegel G, Gerber H, Koch P, et al. The Alzheimer's disease γ-secretase generates higher 42∶40 ratios for β-amyloid than for p3 peptides[J]. Cell Rep, 2017, 19(10):1967-1976.
[2] Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, et al. Alzheimer's disease-associated β-amyloid is rapidly seeded byto protect against brain infection[J]. Neuron, 2018, 99(1):56-63.e3.
[3] Charidimou A, Friedrich JO, Greenberg SM, et al. Core cerebrospinal fluid biomarker profile in cerebral amyloid angiopathy: a meta-analysis[J]. Neurology, 2018. 90(9):e754-e762.
[4] Baker-Nigh A, Vahedi S, Davis EG, et al. Neuronal amyloid-β accumulation within cholinergic basal forebrain in ageing and Alzheimer's disease[J]. Brain, 2015, 138(Pt 6):1722-1737.
[5] Roher AE, Esh CL, Kokjohn TA, et al. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer's disease[J]. Alzheimers Dement, 2009, 5(1):18-29.
[6] Stakos DA, Stamatelopoulos K, Bampatsias D, et al. The Alzheimer's disease amyloid-beta hypothesis in cardiovascular aging and disease: JACC Focus Seminar[J]. J Am Coll Cardiol, 2020, 75(8):952-967.
[7] Chen LD, Zhu WT, Cheng YY, et al. T-cell death-associated gene 8 accelerates atherosclerosis by promoting vascular smooth muscle cell proliferation and migration[J]. Atherosclerosis, 2020, 297:64-73.
[8] Xie B, Shi X, Xing Y, et al. Association between atherosclerosis and Alzheimer's disease: a systematic review and meta-analysis[J]. Brain Behav, 2020, 10(4):e01601.
[9] Stamatelopoulos K, Sibbing D, Rallidis LS, et al. Amyloid-beta (1-40) and the risk of death from cardiovascular causes in patients with coronary heart disease[J]. J Am Coll Cardiol, 2015, 65(9):904-916.
[10] Tublin JM, Adelstein JM, Del Monte F, et al. Getting to the heart of Alzheimer disease[J]. Circ Res, 2019. 124(1):142-149.
[11] Lu DZ, Dong W, Feng XJ, et al. CaMKII(δ) regulates osteoclastogenesis through ERK, JNK, and p38 MAPKs and CREB signalling pathway[J]. Mol Cell Endocrinol, 2020, 508:110791.
[12] Quan GH, Wang H, Cao J, et al. Calycosin suppresses RANKL-mediated osteoclastogenesis through inhibition of MAPKs and NF-κB[J]. Int J Mol Sci, 2015, 16(12):29496-29507.
Aβ1-40 induces inflammation and phenotypic switching via activation of MAPKs signaling pathway in vascular smooth muscle cells
ZHANG Jie, HUANG Xing-xiao, CHI Ju-fang
[,(,),312000,]
To investigate the effects of amyolid β-protein 1-40 (Aβ1-40) on inflammation, viability, migration and phenotypic switching in vascular smooth muscle cells (VSMCs), and to analyze the underlying mechanisms.The VSMCs were treated with Aβ1-40 at different concentration gradients for appropriate time. CCK-8 and Transwell assays were performed to evaluate the viability and migration ability of VSMCs. The levels of inflammatory factors including interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), phenotypic switching-related proteins including α‑smooth muscle actin (α‑SMA), osteopontin (OPN) and Krüppel‑like factor 4 (KLF4), and mitogen-activated protein kinases (MAPKs) signaling pathway-related proteins including p-p38 MAPK, p-ERK1/2 and p-JNK were determined by Western bolt.After Aβ1-40 treatment, the levels of inflammatory factors IL-1β and TNF-α in the VSMCs were significantly increased (<0.05), and the expression of phenotypic switching-related proteins was altered, as indicated by down-regulation of α‑SMA and up-regulation of OPN and KLF4 (<0.05). Treatment with Aβ1-40 within a certain concentration range promoted the viability and migration of the VSMCs. In addition, the protein levels of p-p38 MAPK, p-ERK1/2 and p-JNK were significantly increased by Aβ1-40 treatment (<0.05). Furthermore, pretreatment with specific inhibitors of MAPKs pathway significantly reduced the levels of IL-1β and TNF-α (<0.05), and inhibited the phenotypic switching, as indicated by up-regulation of α‑SMA and down-regulation of OPN and KLF4 (<0.05).Treatment with Aβ1-40 induces the inflammation and phenotypic switching in VSMCs via activation of MAPKs signaling pathway.
Amyolid β-protein 1-40;Vascular smooth muscle cells; Inflammation; Phenotypic switching; MAPKs signaling pathway
R749.1+6; R363.2
A
10.3969/j.issn.1000-4718.2020.11.007
1000-4718(2020)11-1966-06
2020-04-27
2020-07-01
国家自然科学基金资助项目(No.81873120);浙江省医药卫生科研资助项目(No.2018KY172)
Tel: 0575-88228888; E-mail: chijufang@zju.edu.cn
▲共同第一作者:并列第1作者
(责任编辑:宋延君,罗森)
猜你喜欢
山东医药(2022年26期)2023-01-06
河北果树(2021年4期)2021-12-02
天津医科大学学报(2021年1期)2021-01-26
食品安全导刊(2020年21期)2020-12-03
医药前沿(2020年20期)2020-11-10
湖南农业科学(2020年1期)2020-04-18
中国CT和MRI杂志(2020年3期)2020-03-27
中国临床医学影像杂志(2019年4期)2019-06-18
河北农业科学(2019年6期)2019-03-21
食品安全导刊(2019年30期)2019-01-05
