
基因组编辑技术及其在合成生物学中的应用
2020-11-18 07:14曹中正张心怡徐艺源周卓魏文胜1
合成生物学 2020年4期
曹中正,张心怡,徐艺源,周卓,4,5,6,魏文胜1,,4,5,6
(1 北大-清华生命科学联合中心,北京 100871;2 北京大学前沿交叉学科研究院,北京 100871;3 北京大学生命科学学院,北京 100871;4 北京大学生物医学前沿创新中心,北京 100871;5 北京未来基因诊断高精尖创新中心,北京100871;6 北京大学基因组编辑研究中心,北京 100871)
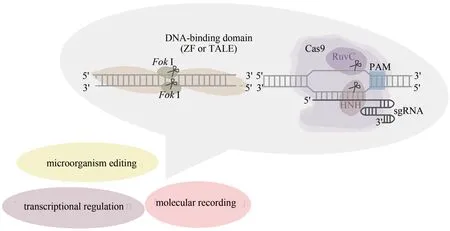
随着人类基因组计划的完成和测序数据的不断积累,研究者们已经掌握了海量的DNA 遗传信息,但是这些信息所代表的生物学意义却有待进一步的挖掘。近年来,以基因组编辑为代表的新型生物技术正在飞速发展。这种简单易行的技术使得研究者们可以对基因组或者基因转录后产物进行编辑,从而大大提高了科学家们解码基因功能以及调控基因表达的能力。基因组编辑这一革命性技术的发现与完善,必将对人类的基础科学研究、工农业生产以及生命健康等诸多领域产生深远的影响。本文将着重介绍以CRISPR 系统为代表的新型基因组编辑技术,并对该技术在合成生物学中的应用进行阐述。
1 基因组编辑技术
基因组编辑技术,是指一种对目标基因进行编辑或修饰的基因工程技术。目前,应用最为广泛的主要有以下3种技术:锌指蛋白核酸酶(zincfinger nuclease,ZFN)[1]、类转录激活因子效应物核酸酶(transcription activator-like effector nuclease,TALEN)[2]以及CRISPR/Cas(clustered regularly interspaced short palindromic repeats,CRISPR;CRISPR-associated,Cas)系统[3-5]。
1.1 早期基因组编辑工具
1.1.1 ZFN技术
ZFN 是由锌指蛋白(zinc-finger protein,ZFP)DNA 结合域与核酸内切酶FokI切割结构域融合而成的一种基因编辑工具[1]。锌指是构成锌指蛋白的基本单元,每个锌指大约含有30 个氨基酸,并通过保守的Cys2-His2 残基与锌原子螯合,形成稳定而紧实的β-β-α(N 端-C 端)基序[6-8]。锌指α 螺旋表面的氨基酸残基与DNA 双螺旋大沟中的碱基通过相互作用进行识别,每个锌指能够识别3个碱基。因此,将不同的锌指进行串联就可以设计出识别特定DNA序列的锌指蛋白[9-10]。
FokI是一种ⅡS 型限制性核酸内切酶[11],与识别特定DNA 序列的锌指蛋白融合后,可构成一个锌指蛋白核酸酶的单体[1]。只有当两组锌指蛋白核酸酶的单体分别靶向DNA 的正反两条链,并在基因组上具备适宜的间隔时(通常是5~7 bp),FokI 才能够形成具有切割活性的二聚体[12-13]。ZFN 产生双链断裂(double-stranded break,DSB)后, 会激活细胞内非同源末端连接(nonhomologous end joining,NHEJ)[14]或同源重组(homologous recombination,HR)[15]修复途径,从而实现对特定基因的编辑。但ZFN 设计成本昂贵,制备复杂,且容易受到上下文效应(context effect)的影响,严重限制了该技术的推广与应用[8]。
1.1.2 TALEN技术
类转录激活因子效应物(transcription activator-like effector,TALE)是一种来自于植物致病菌黄单胞杆菌(Xanthomonas)中的蛋白[16]。TALE 蛋白N 端是转运信号(translocation signal)[17], C 端含有核定位信号(nuclear localization signal)[18]与转录激活结构域(activation domain)[19],中间部分则是具有特异性识别和结合DNA 功能的结构域[20]。这些DNA 结合结构域由33~35 个氨基酸组成的重复序列串联而成,在这些重复序列中,第12、13 位氨基酸是高度可变的,被称为重复可变双残基(repeat variable di-residue,RVD)[21-22]。RVD 的不同决定了TALE识别碱基的特异性,例如NI、HD、NG和NN 四种RVD 分别对应识别A、C、T 和G 四种碱基[21]。与ZFN 类似,研究者们将不同TALE 蛋白串联后与FokI 融合形成TALEN,两个TALEN 单体分别靶向DNA 的两条链即可达到基因编辑的目的[2]。由于TALEN 的制备比较复杂,其应用也有着一定的限制。
1.2 CRISPR/Cas系统
1.2.1 CRISPR/Cas系统的发现与发展
CRISPR/Cas 系统是细菌和古生菌中一种抵御外源遗传物质入侵的适应性免疫机制。1987 年,科学家们最早在大肠杆菌中发现了间隔串联重复序列[23],随后的研究发现这样的重复序列广泛存在于细菌与古生菌中[24]。2007 年,Barrangou等[25]提供了CRISPR 作为免疫系统抵抗噬菌体侵染的首个直接证据。2010 年,Garneau 等[26]首次证实了crRNA(CRISPR RNA)可以在体内介导CRISPR 系统切割双链DNA。2011 年,Siksnys 课题组[27]将嗜热链球菌的CRISPR 系统转移到大肠杆菌中,发现这种异源系统可以抵抗质粒转化和噬菌 体 侵染。2012 年,Doudna 和Charpentier 课题组[4]以及Siksnys 课题组[28]证实,体外重构的CRISPR 系统可以对双链DNA 进行切割。2013 年,Zhang 课题组[3]和Church 课 题 组[5]成 功 运 用CRISPR 系统,在哺乳动物细胞中实现内源基因的编辑。至此,CRISPR 系统开始发展成为快捷高效的基因组编辑工具。
CRISPR/Cas 通常由一系列CRISPR 相关(CRISPR-associated,Cas)基因与CRISPR 阵列组成。CRISPR 阵列由一系列高度保守的正向重复序列和序列特异的间隔序列组成。目前CRISPR 系统可以被分为2大类和6小型[29-31],1类系统包括I型、Ⅲ型和Ⅳ型,2 类系统包括Ⅱ型、V型和Ⅵ型,每种类型使用一组独特的Cas蛋白和crRNA来行使功能[32]。本文作者将以Ⅱ型CRISPR 系统产脓链球菌的SpCas9(Streptococcus pyogenesCas9)蛋白为例,介绍其具体的作用机理。
1.2.2 CRISPR/Cas9系统的作用机制
CRISPR 介导的防御机制可分为3 个阶段:适应、表达与干扰[33]。在适应阶段,系统对入侵质粒或噬菌体的DNA 片段进行选择和切割[25],在Cas1-Cas2 复合体的帮助下,将其整合成新的间隔序列[33]。间隔序列的选择由原间隔区相邻基序(protospacer adjacent motif,PAM)引导,不同物种来源的CRISPR/Cas 系统具有不同的PAM,同时PAM 对于系统区分自身序列和外源序列具有至关重要的作用[34]。在表达阶段,系统将CRISPR阵列转录成长的前体CRISPR RNA(pre-crRNA),同时转录出与pre-crRNA 中重复序列互补配对的反式激活crRNA (trans-activating crRNA, tracrRNA)。pre-crRNA 与tracrRNA 的互补配对能够触发体内RNase Ⅲ等核酸酶的切割机制,产生一系列间隔序列不同的成熟crRNA[4,35]。在干扰阶段,Cas9 蛋白与tracrRNA-crRNA 形成的复合物在外源基因组上寻找PAM 序列,并在PAM 序列处停留与识别。一旦间隔序列能够与外源基因组上的序列完全互补配对,Cas9 蛋白就会对基因组位点进行切割,从而达到降解外源遗传物质的目的[36]。
1.2.3SpCas9的结构及其系统优化
SpCas9 的结构被分为α 螺旋识别区(recognition lobe, REC) 和 核 酸 酶 区(nuclease lobe,NUC)[37]。前者是Cas9 蛋白家族中序列保守度最低的区域,负责RNA-DNA的识别,而核酸酶区则包含了HNH、RuvC 以及负责与PAM 识别的PI 结构域。RuvC 和HNH 是两个核酸酶结构域,其中HNH 以独立的形式存在,而RuvC 则被分成3 个亚结构域,RuvC I位于SpCas9 的N 端,RuvC Ⅱ和Ⅲ排列在HNH结构域的两端。HNH结构域对sgRNA的互补链进行切割,而RuvC结构域则对sgRNA的非互补链进行切割(图1)。两个区之间通过富含精氨酸的桥螺旋(bridge helix,BH)连接起来,该部分与sgRNA有直接的相互作用。
为了更便捷地使用CRISPR/Cas9 系统,研究人员对其进行了一些优化。Jinek 等[4]通过一段GAAA序列将crRNA的3ʹ端与tracrRNA的5ʹ端融合在一起,形成了单链引导RNA(single guide RNA,sgRNA),在不影响效率的同时简化了系统组分(图1)。此外,Zhang 课题组[3]对SpCas9 蛋白进行密码子优化,并加入核定位信号,提高了SpCas9 蛋白转运到哺乳动物细胞核内的效率。有研究表明,sgRNA 在哺乳动物细胞内的表达水平较低,限制了CRISPR/Cas9 系统的应用[38]。因此Chen 等[39]对sgRNA 的骨架结构进行了优化:他们将sgRNA 茎环结构中第4 个连续的U 替换为A,避免了Pol Ⅲ启动子的提前终止;同时他们将sgRNA 骨架上的发夹结构延伸了5 对碱基,以便SpCas9 与sgRNA 的结合。基于以上的优化,CRISPR/Cas9 系统只需要导入Cas9 与sgRNA 即可实现高效的基因组编辑。
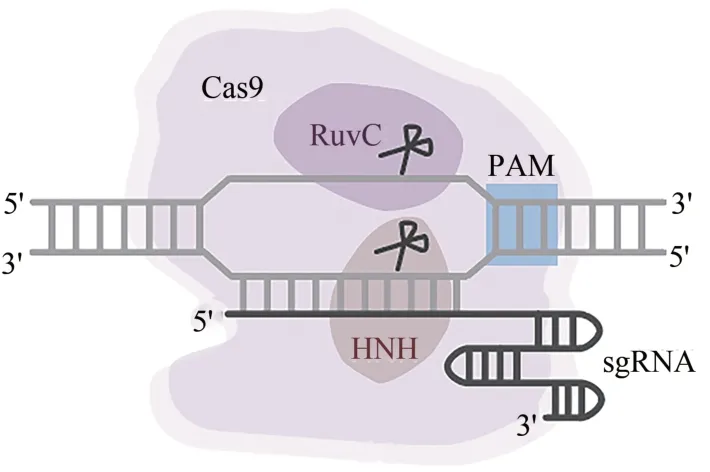
图1 CRISPR/Cas9系统介导基因编辑Fig.1 Schematic of the CRISPR/Cas9 mediated genome editing
1.2.4 多种CRISPR/Cas系统
由于1 类CRISPR 系统需要多个Cas 蛋白形成复合物才能发挥功能,而2 类系统只需要单个Cas蛋白即可对靶向序列进行切割[29],因此2 类系统被广泛应用于各项研究中。除了SpCas9,目前还有多种系统正处于开发和应用的过程中。SaCas9(Staphylococcus aureusCas9)是来自金黄色葡萄球菌的Cas9 蛋白,其作用机理和编辑效率与SpCas9类似,识 别NNGRRT 的PAM 序列[40]。SaCas9 蛋白较小,只有1053 个氨基酸,可以和sgRNA 一起被装载进单个腺相关病毒(adeno-associated virus,AAV)载体中,因此在基因治疗的临床应用上具有优势。2015 年,Zetsche 等[41]报道了另一种2 类系统Cpf1(也称Cas12a),该系统只需要crRNA 介导即可进行基因组编辑,不依赖于tracrRNA 的产生。Cpf1识别PAM序列为TTN,切割靶向DNA后会产生黏性末端。随后,通过对CRISPR/Cas 基因座进行序列比对分析, Shmakov 等[30]与Abudayyeh 等[42]报 道 了C2c1(Cas12b)、C2c2(Cas13a)等新系统。其中C2c2 是靶向RNA 的系统,通过一条crRNA 介导,能够实现对单链RNA的切割。相信在不远的将来,具有新型应用特点的CRISPR系统会不断被发现。
1.2.5 CRISPR/Cas延展技术
随着研究的深入,人们对CRISPR 结构和功能的认知越来越清晰,多种基于CRISPR 系统的新工具也应运而生。2013 年,Qi 等[43]对Cas9 核酸酶结构域进行突变(H840A,D10A),产生了只有结合功能但不具有切割活性的dCas9(dead Cas9),实现了利用CRISPR 进行基因表达调控的目的,他们将这一系统命名为 CRISPRi (CRISPR interference)。该技术通过靶向基因的转录起始位点(transcriptional starting site,TSS),干扰RNA聚合酶或转录因子与DNA 的结合以及转录的延伸,从而达到抑制基因表达的目的。在此基础上,Gilbert等[44]将dCas9 与转录抑制因子KRAB 融合,提高了CRISPRi 的效率,有效抑制了内源基因的表达。此外,研究人员将dCas9与招募转录激活因子的蛋白例如VP64 或p65 激活结构域(p65AD)融合,开发出CRISPRa(CRISPR activation)系统,可以高效上调基因的表达[44-46]。
如果将调节转录的结构域替换成其他功能的蛋白,则可以产生多种不同的工具。例如,通过将dCas9 与组蛋白去甲基化酶LSD1 进行融合,Kearns 等[47]建立了一种可以检测远端顺式调控元件的方法。Hilton 等[48]将dCas9 与乙酰转移 酶p300 进行融合,开发了可以在表观修饰水平上调节基因表达的工具。在活细胞成像领域dCas9系统同样大放异彩,例如Chen 等[39]将EGFP 与dCas9蛋白融合在一起,利用sgRNA 介导,成功在活细胞中实现了端粒重复元件与编码基因的成像,促进了自然状态下染色体构象和动力学的研究。
2016 年,多个课题组开发了可以将胞嘧啶转换为胸腺嘧啶的碱基编辑器(cytosine base editor,CBE),在不引入DSB 的情况下,实现了精准的单碱基编辑[49-51]。经过一系列的优化[52],CBE 目前常用的形式是将胞嘧啶脱氨酶与Cas9 切口酶(nickase Cas9,nCas9)融合,通过sgRNA 靶向到特定序列,脱氨酶将编辑窗口内单链DNA 上的胞嘧啶脱氨成尿嘧啶,nCas9 则在与sgRNA 互补的DNA 链上进行切割,造成单链断裂,激活细胞内DNA 修复机制,促使细胞以被脱氨后的单链为模板进行修复,将原有的C-G 碱基对替换为T-A。同时融合表达的尿嘧啶DNA 糖苷酶抑制剂(uracil DNA glycosylase inhibitor,UGI)能够抑制碱基切除修复途径,提高编辑效率。2017 年,Liu 课题组[53]进一步扩大了碱基编辑系统的类型,通过将人工进化的腺嘌呤脱氨酶与nCas9融合,开发出能将腺嘌呤转换为鸟嘌呤的腺嘌呤碱基编辑器(adenine base editor,ABE)。腺嘌呤脱氨后变成次黄嘌呤,在下一次DNA 复制过程中被识别为鸟嘌呤,实 现A 到G 的替 换。2019 年,Liu 课 题 组[54]又报道了Prime Editor 技术,将逆转录酶融合在nCas9 的C 端,同时在sgRNA 的3ʹ端延伸出一段序列作为逆转录的引物和模板,实现了任意碱基之间的替换以及特定碱基序列的插入与删除。
1.2.6 CRISPR/Cas系统的应用
CRISPR/Cas9系统作为一种高效的基因组编辑技术,目前已成功应用于酿酒酵母[55]、线虫[56]、果 蝇[57]、斑 马 鱼[58]、拟 南 芥[59]、水 稻[60]、小鼠[61]、大鼠[62]以及多种人类细胞系[3,5]中,同时其在高通量筛选、基因治疗、农作物育种及性状改良等方面也有着广泛的应用。
CRISPR/Cas9系统在建立混合文库型高通量筛选策略之后[63-66],被迅速运用于多种细胞系中必需基因的筛选[67]以及非编码调控元件的筛选[68-72],这些研究极大促进了科学家们对基因功能的理解。单细胞测序技术与CRISPR 高通量筛选技术的结合,大大拓宽了高通量筛选的适用场景[73-77]。研究者们将等温扩增技术与CRISPR/Cas13a、CRISPR/Cas12a 等系统结合,开发了一系列快速灵敏的核酸检测技术[78-82],可以在短时间内检测病原物的存在,例如寨卡病毒、登革热病毒等。此外,利用CRISPR/Cas9 与第3 代纳米孔测序技术,研究人员实现了在起始基因组量很少的情况下,对DNA 突变、区域修饰以及结构改变等的检测[83]。
作为一种特殊的基因治疗方式,CRISPR 系统可以通过基因组编辑对细胞进行工程改造,从而治疗多种疾病,例如单基因遗传病的突变校正、嵌合抗原受体(chimeric antigen receptor,CAR)T细胞疗法的改进以及再生医学的应用等[84]。例如,β-地中海贫血病的治疗是通过腺相关病毒将CRISPR/Cas9系统与同源重组模板递送到病人造血干细胞中,实现HBB基因突变位点的校正[85]。Xu等[86]通过CRISPR/Cas9 系统对成体造血干细胞的CCR5基因进行编辑,并回输到患有艾滋病和白血病的患者体内,成功缓解了两种疾病的症状。此外,利用CRISPR/Cas9 技术编辑内源基因,可以最大限度地减弱T细胞受体或人类白细胞抗原介导的排异反应,产生更高效的CAR-T 细胞[87-89]。在作物育种与性状改良方面,CRISPR 系统也发挥了巨大的作用,研究人员已经实现了作物产量相关性状的改良[90-92]、营养质量的提高[93-95]以及抗逆抗病能力的增强[96-98]。
2 基因组编辑技术在合成生物学中的应用
合成生物学是一门新兴的交叉学科,主要利用工程学的思维在生物学领域进行应用,旨在提高解码和重编程生物学系统的能力[99]。合成生物学能够改造和优化已有的自然生物体系,或者重新设计合成具有特定功能的人工生物体系,实现在化工、能源、医药、环境等领域的规模化应用。在合成生物学的标准化以及模块化等诸多过程中,基因组编辑技术起着重要的作用。下面将介绍基因组编辑技术在合成生物学中的应用,着重以CRISPR系统为例(图2)。
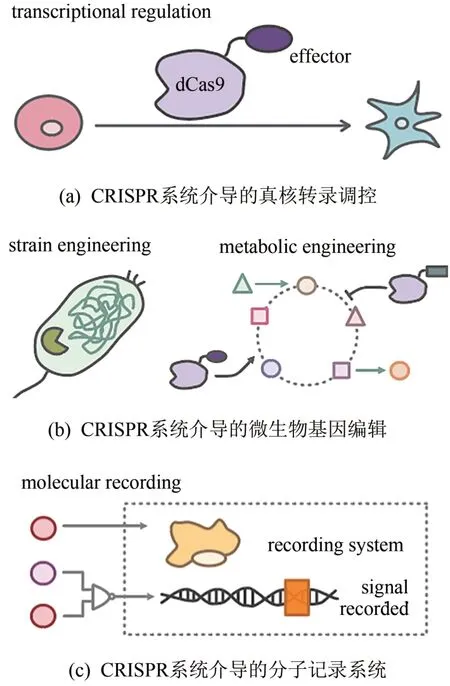
图2 CRISPR系统在合成生物学中的应用Fig.2 Applications of the CRISPR system in synthetic biology
2.1 基因组编辑技术介导的真核转录调控
基因组编辑技术可以进行精准的转录调控,因此被广泛应用于基因动态过程的调控以及细胞命运的操纵。
Rivenbark 等[100]将ZFP 与DNA 甲 基 转 移 酶DNMT3a融合,使得Maspin和SOX2基因的启动子区域产生甲基化修饰,在乳腺癌细胞中实现了长期稳定的基因表达下调。Khalil 等[101]利用人工合成的ZFP 与多种转录因子融合,构建了正交合成的转录因子文库,通过改变不同的转录因子输入信号,可以在酵母中研究单个转录因子以及转录调控网络的功能。Keung 等[102]将223 个酵母染色质调节因子与ZFP 融合,通过研究不同时空组合下染色质调节因子所产生的转录行为,解析了染色质调控的复杂性,为染色质介导的转录调控提供了新的工具。Maeder 等[103]利用TALE 与TET1羟化酶的融合蛋白,实现了启动子区域CpG 位点的去甲基化,从而调节了内源基因的表达。Mendenhall 等[104]将TALE 与组蛋白去甲基化酶LSD1 融合,通过在靶位点上去除增强子相关的染色质修饰,下调近端基因的表达。Konermann等[105]将TALE 与光敏蛋白CRY2 及其互作蛋白CIB1 融合,开发了一套基于光诱导的双杂交转录效应物系统LITE,成功在小鼠神经元细胞以及大脑中实现了可逆的内源基因表达及染色质表观修饰调控。Zhang 等[106]通过融合TALE 与VP64,在293FT 细胞中实现了内源基因SOX2与KLF4的上调表达。
Chavez 等[107]通过dCas9 与转录激活结构域VP64-p65-Rta 的顺序组合,建立dCas9-VPR 系统,成功在体内激活内源的编码基因和非编码基因,促使人诱导多能干细胞向神经元细胞的分化。Tanenbaum 等[108]通过在dCas9 的C 端融合表达多个肽簇,同时表达该肽簇对应抗体与VP64 的融合蛋白,建立了Suntag 系统,高效地实现了CXCR4等基因的激活调控。Konermann 等[109]通过在sgRNA 上拼接适配体,并通过表达能够识别RNA适配体的转录激活因子融合蛋白,建立了SAM 系统,成功实现了内源基因和非编码基因的激活,同时完成了BRAF 抑制剂的全基因组筛选。Chakraborty 等[110]利用dCas9-VP64 系统上调小鼠MYOD1基因的表达,成功使得小鼠胚胎成纤维细胞分化为骨骼肌细胞。Kearns 等[111]利用dCas9-VP64 系统上调了指征人胚胎干细胞中内胚层分化的SOX17基因的表达,同时也利用CRISPRi 系统对人胚胎干细胞OCT4基因和NANOG基因进行抑制,从而影响了干细胞的多能 性。Liu 等[112]将dCas9 与DNMT3a 或Tet1 融合,通过催化甲基化或者去甲基化,在体内外均能够实现基因的表达抑制或激活。Kwon等[113]通过将dCas9 与组蛋白去乙酰化酶HDAC3融合,发现可以对内源基因实现转录抑制,并且抑制效率与sgRNA 呈现剂量依赖效应。诱导型的转录调控系统可以响应光或小分子等外界信号,在特定信号存在时,启动转录调控,这一手段对了解基因的动态网络十分重要[114-117]。Qi 课题组[117]报道了诱导型的正交转录调控系统,在同一细胞内实现了复杂的转录调控。基于dSpCas9 与dSaCas9 的正交性, 他们利用dSpCas9-KRAB 和dSaCas9-VPR 在抑制EGFP 报 告基因表达的同时上调了mCherry 报告基因的表达。此外,他们将脱落酸诱导系统和赤霉素诱导系统与转录调控系统组合,产生多种AND、OR、 NAND 及NOR 的 布 尔 逻 辑 门。 同 时CRISPR 系统介导的真核转录调控在遗传性疾病的临床治疗中也发挥着重要的作用。Liao 等[118]通过对SAM 系统进行优化,利用AAV 递送靶向序列较短的sgRNA 和野生型的Cas9 进入杜氏肌营养不良的小鼠中,成功上调了DMD基因的表达,显著改善了小鼠的肌肉功能。Liu 等[119]利用dCas9-Tet1 融合蛋白,在脆性X 染色体综合征病人的诱导多功能干细胞中,成功激活了FMRP基因的转录表达,这种激活作用能够持续至少两周时间,并且没有检测到明显的脱靶作用。Moreno 等[120]利用两个AAV 载体,将CRISPRi系统递送至色素性视网膜炎小鼠模型的视网膜下空间,成功实现了对Nr1基因的抑制,调控了视杆细胞向视锥细胞分化,达到了治疗的效果。
2.2 基因组编辑技术介导的微生物基因编辑
微生物一直是许多抗生素类药物和活性天然产物的重要合成来源,但是这些次级代谢产物在衍生品开发和规模化制备等方面都存在瓶颈,需要对特定的基因或者同时对合成通路里面的多个基因进行编辑,从而达到改良菌种的目的。
Zhang 课 题 组[121]利用TALEN 在 酿酒酵母(Saccharomyces cerevisiae)中对乙醇脱氢酶ADH2进行敲除,从而大幅度提高了酒精的产量。而后该课题组[122] 在纤维素堆囊菌(Sorangium cellulosum)中,利用TALE-VP64 与dCas9-VP64系统,使埃博霉素的产量得到了不同程度的提升。研究者们将CRISPRi 系统用于谷氨酸棒状杆菌(Corynebacterium glutamicum),可以提升L-赖氨酸的效价,增加L-谷氨酸的产量[123-124]。Wu 等[125]在大肠杆菌(Escherichia coli)中应用CRISPRi 系统对糖酵解途径、TCA 循环、脂肪酸合成途径进行调控,实现了类黄酮的增产。此外,Schwartz等[126]在耶氏解脂酵母(Yarrowia lipolytica)中利用CRISPRa 系统提高β-葡糖苷酶的表达,获得了以纤维二糖为碳源的改良菌株,拓宽了这一工程菌的适用范围。
研究者们也利用CRISPR 系统进行多基因的编辑,希望能够快速高效地获得目标菌株。Cobb等[127]开发出pCRISPomyces 方法,成功地在变铅青链霉菌(Streptomyces lividans)中同时敲除了redN和actVA-ORF5基因,并证实该方法在白色链霉菌(Streptomyces albus) 和产绿色链霉菌(Streptomyces viridochromogenes) 中具有67%~100%不等的敲除效率。Jia 等[128]通过对环状链霉菌(Streptomyces rimosus)的zwf2和devB基因进行敲除,使抗菌药物土霉素的产量提高了36.8%。Zhang等[129]在酿酒酵母中利用gRNA-tRNA串联的阵列,能够同时靶向8个目的基因,他们将此方法应用于简化酵母脂质代谢网络,成功使得游离脂肪酸的产量提高了30倍。Li等[130]利用CRISPR/Cpf1系统在天蓝色链霉菌(Streptomyces coelicolor)中实现了一个或者多个基因的敲除,效率最高可达95%。Tong 等[131]利用碱基编辑技术开发出CRISPR-BEST 系统,成功地在放线菌黄色霉素合成通路的kirN基因中引入提前的终止密码子,通过结合Csy4 他们还实现了多个位点的同时编辑。另外,通过对多个基因进行快速敲除,能够鉴定未知的生物合成途径。例如在旱地小单孢菌(Micromonospora chersina)中,通过敲除8个不同的基因簇,Cohen 等[132]成功鉴定出聚酮合酶DynE8同时参与烯二炔和蒽醌的生物合成。除此之外,CRISPR/Cas9系统还可以将异源的启动子定点整合至基因的上游,从而激活基因的表达。例如,Zhang 等[133]利用该策略在玫瑰链球菌(Streptomyces roseosporus)得到具有抗真菌活性的物质、 在委内瑞拉链霉菌(Streptomyces venezuelae)中得到具有抗疟原虫活性的物质以及在产绿色链霉菌中得到新型的色素类产品。
2.3 基因组编辑技术介导的分子记录系统
基因组编辑技术的出现使得人们可以在活细胞中动态地更改遗传信息,并利用基因组DNA 的强大承载力对信息进行存储[134]。基因组编辑技术通过响应光、温度或金属离子等对分子记录器元件进行控制,可以实现对特定信号的活细胞记录。分子记录系统在研究细胞分化、免疫细胞的发育、疾病及环境监测等领域有着重要应用。
依赖于CRISPR 系统的分子记录器可以分为3 类:①碱基编辑器介导的分子记录系统;②Cas9核酸酶及sgRNA 介导的分子记录系统;③Cas1-Cas2蛋白介导的分子记录系统。
碱基编辑器介导的分子记录系统是利用dCas9(或nCas9)和脱氨酶的融合蛋白,在sgRNA 的引导下,对特定位点进行脱氨,达到改写的目的。Liu 研 究 组[135]开 发 的CAMERA 技 术 和Lu 研 究组[136]开发的DOMINO 技术均是利用可调控的碱基编辑器,在被检测信号存在时,完成位点特异性的编辑,并通过荧光信号的变化或该位点编辑比例的变化指征事件的发生。这一类系统由于产生精准的突变类型,可以被用于模拟记录、数字记录和各类复杂计算中。
Cas9 核酸酶及sgRNA 介导的分子记录系统是指,通过NHEJ修复途径在靶位点产生的丰富突变作为每一个细胞特有的条形码,对特定的信号或细胞的发育过程进行记录,以完成谱系追踪。Shendure 研究 组及合 作 者[137]开发了GESTALT(genome editing of synthetic target arrays for lineage tracing)技术,通过细胞之间突变模式的不同来区分它们的世代关系,并对斑马鱼的细胞进行谱系重构,发现大多数成体斑马鱼的细胞均来自于少数胚胎祖细胞。Cai 研究组及合作者[138]报道了MEMOIR(memory by engineered mutagenesis with opticalin situreadout)技术,在CRISPR/Cas9 系统产生突变后利用单分子RNA 荧光杂交(smFISH)技术,完成了胞内发生事件的记录以及胚胎干细胞的谱系重构。除对外源的靶位点进行突变以外,sgRNA 表达序列本身也可以被当作靶位点[139-140]。Lu 研究组[140]开发了mSCRIBE (mammalian synthetic cellular recorders integrating biological events)技术,将sgRNA 表达序列作为靶位点,利用sgRNA 自切割所产生的多种突变作为条形码,完成了NF-κB 通路激活和LPS 介导的炎症反应的记录。
Cas1-Cas2 蛋白介导的分子记录系统是利用CRISPR 系统的间隔序列获取机制,将外源DNA整合进前导序列中,以记录特定序列的存在。间隔序列的顺序,也指征着该特定序列被Cas1-Cas2蛋白获取的先后次序[141-143]。Church 研究组[142]利用Cas1-Cas2 介导的间隔序列获取系统,对黑白图像的像素值和一部数字电影进行了编码,并将其记录在细菌中。Wang 研究组[143]开发了TRACE(temporal recording in arrays by CRISPR expansion)系统,以生物录音带的形式将外界信号记录储存下来,同时他们也尝试了对铜、海藻糖和岩藻糖这类代谢物进行记录。
3 展 望
合成生物学是一个高度交叉的研究领域,涉及基础的生物学研究、工程技术和计算机建模等诸多方面,因此合成生物学的突破依赖于各个学科的协调发展。基因组编辑技术的出现加速了合成生物学的发展,但是该技术仍然存在一些问题以及改善的空间,同时该技术在合成生物学领域的应用也有待进一步的开发。
初代的基因组编辑技术ZFN 和TALEN,虽说在构建上略显烦琐,但是在某些应用场景中仍然具有独特的优势,例如对真核细胞线粒体基因组的编辑。而目前以CRISPR/Cas9 为代表的新型基因组编辑工具最为研究者们所关注的问题有三点:一是拓宽可编辑的范围;二是减少脱靶效应;三是提高递送效率。由于CRISPR 系统主要来源于微生物,研究者们可以通过生物信息学的方法从环境微生物(例如极端环境微生物)的测序数据中发掘出具有不同特性的新型系统,利用此方法拓宽可编辑的范围是未来的一个重要研究方向。同时研究者们也可以对已有的系统进行优化,使其具有识别更广阔PAM 的能力[144-148]。这些方法虽然一定程度上减弱了PAM 限制,但是大多时候也会降低原始PAM 的限制作用,从而加剧脱靶效应,在使用时应当按照实际需求选择适宜的突变体。对于减少脱靶效应,目前研究者们可以通过结构优化或者随机突变的形式获得高保真的突变体[149-151]。但是这些突变体均不完美,都是以牺牲打靶效率为代价来降低脱靶效应,因此继续优化发展真正意义上的高保真突变体将是未来的研究重点。此外,小型Cas 蛋白的发现[152-153]或现有Cas 蛋白的长度优化[154]对于提高AAV 等病毒载体的递送效率具有重要的意义。
虽然基因组编辑技术在合成生物学领域已经有了诸多的应用,但是依然存在较大的改善空间。首先在基因表达调控方面,发展多种正交的系统可以实现对不同基因不同方向的表达调控,这对于基因网络的研究具有十分重要的作用。同时,发展可控的表达调控系统(例如小分子诱导或者光控)以及运用布尔逻辑门也有助于对目的基因的精准调控。其次在微生物编辑方面,目前绝大多数情况下只能通过效率较低的同源重组对双链断裂进行修复[155],极大限制了该技术的应用。未来可以通过碱基编辑器和引导编辑器(PE 系统)等不产生双链断裂的技术提高编辑效率或者结合CRISPR 系统与重组酶等实现高效的插入和删除,从而达到改良菌种、优化更多生物合成路径的目的。此外,合成生物学利用CRISPR 系统已经能够初步实现信息的存储和读取,但在效率、容量和稳定性方面仍有较大的提升空间,希望将来可以将生物存储应用于更多的场景。
综上所述,基因组编辑技术的发展能够极大促进合成生物学的应用,同时合成生物学又能够指导和优化现有的基因组编辑工具,两者相辅相成,必将在生物医药、化工生产、环境能源等诸多领域取得突破性的进展。
猜你喜欢
军事文摘(2022年16期)2022-08-24
湖北农业科学(2022年11期)2022-07-18
河北农业大学学报(2022年2期)2022-04-26
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
实用肿瘤学杂志(2020年4期)2020-12-08
教学考试(高考生物)(2020年6期)2020-11-23
中西医结合肝病杂志(2020年2期)2020-10-27
食品与生物技术学报(2020年8期)2020-01-06
学苑创造·B版(2019年5期)2019-06-14
