
Bruton酪氨酸激酶在对乙酰氨基酚诱导的小鼠肝损伤中变化及意义
2020-09-09 08:25刘晓庆鲁桓兵刘瑞雪宋育林
安徽医科大学学报 2020年8期
刘晓庆,鲁桓兵,刘瑞雪,宋育林
对乙酰氨基酚 (acetaminophen,APAP)是目前普遍使用的解热镇痛剂,过量使用则可导致严重的肝功能损害甚至急性肝衰竭乃至死亡,是药物性肝损伤及肝衰竭的常见原因。APAP所致肝损伤发病机制复杂,涉及多种机制,可能与活性代谢产物的形成、氧化应激、内质网应激、自噬、无菌性炎症等有关[1],确切机制目前仍不明确。核苷酸结合寡聚域样受体蛋白 3(nucleotide-binding oligomerization domain-like receptor protein 3, NLRP3)是肝脏中研究较多的炎性小体,其活化后可引起白细胞介素(interleukin, IL)-1β、IL-18等炎性因子的释放,参与APAP诱导的肝损伤[2]。Bruton酪氨酸激酶(Bruton's tyrosine kinase,Btk)是胞浆非受体酪氨酸蛋白Tec家族成员之一,对B淋巴细胞的增殖、分化和凋亡起着重要的调控作用[3]。近期有文献报道[4]Btk是先天免疫炎性反应中的关键因子,可通过调控核因子κB (nuclear factor κB, NF-κB) 引起NLRP3的激活,也可直接调控NLRP3的激活。已有研究[5]表明Btk参与缺血再灌注性肝损伤,抑制Btk可减轻肝脏的炎性损伤,然而其在APAP诱导的肝损伤中的作用及机制目前未见相关文献报道,该研究重点研究APAP诱导的肝损伤中Btk、磷酸化Btk(phosphorylated Btk,p-Btk)、NF-κB、NLRP3、IL-1β、肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α) 的动态变化,从而探讨Btk在APAP肝损伤中的重要作用及机制。
1 材料与方法
1.1 实验动物54只SPF级健康成年的C57BL/6J雄性小鼠,鼠龄6~8周,体质量18~24 g,购自斯贝福(北京)生物技术有限公司,动物许可证号:SCXK(京)2016-0002。
1.2 实验材料与仪器APAP(纯度大于99%)购自美国MedChemExpress公司;小鼠TNF-α、IL-1β elisa试剂盒购自武汉基因美生物科技有限公司;Btk抗体[Btk(D3H5)Rabbit mAb×8547]、p-Btk抗体均购自美国Cell Signaling公司;NLRP3抗体购自英国Abcam公司;NF-κB抗体购自英国Abcam公司;RIPA裂解液、SDS-PAGE (5×) 蛋白上样缓冲液均购自上海碧云天生物技术有限公司;全自动生化分析仪购自瑞士RocheModular 公司;紫外分光光度计购自上海菁华科技仪器有限公司产品;成像系统购自美国阿尔法公司产品。
1.3 方法
1.3.1实验分组与方法 适应性饲养1周后,54只小鼠随机分为9组,每周6只,分别为对照组(APAP 0 h组)、APAP 0.5 h组、APAP 1 h组、APAP 3 h组、APAP 6 h组、APAP 12 h组、APAP 24 h组、APAP 36 h组、APAP 48 h组,所有小鼠禁食不禁水12 h后,APAP 0.5 h组、APAP 1 h组、APAP 3 h组、APAP 6 h组、APAP 12 h组、APAP 24 h组、APAP 36 h组、APAP 48 h组小鼠分别单剂量腹腔注射APAP 300 mg/kg,对照组小鼠腹腔注射0.9%生理盐水同等体质量APAP组小鼠。所有小鼠自由进食水,明暗各12 h,温度20~25 ℃,相对湿度(50±5)%。所有小鼠均自由进食标准饲料。分别在相应的时间小鼠处死前称量小鼠体质量,用10%的水合氯醛(4 ml/kg)腹腔注射麻醉后摘除小鼠眼球收集血液,断头处死。全血标本于4 ℃冰箱静置过夜后,3 500 r/min离心15 min留取上清液,-20 ℃冻存待测。剪取部分肝左叶用10%福尔马林缓冲液固定行肝脏病理学检查,其余肝脏组织冻存于液氮中,随后转移至-80 ℃ 冰箱储存待测。
1.3.2血清丙氨酸氨基转移酶(alanine aminotransferase,ALT)、天门冬氨酸氨基转移酶(aspartate aminotransferase,AST)活性检测 全自动化生化分析仪检测ALT、AST活性。
1.3.3肝组织病理学观察 经10%福尔马林缓冲液固定的肝脏标本,常规脱水,石蜡包埋,切片(厚5 μm),HE染色,光学显微镜下观察肝组织病理形态学改变。采用Image-Pro Plus 6.0高清晰图像分析系统进行分析。计算肝脏坏死面积百分比=(阳性细胞所占面积/整个视野总面积)×100%。
1.3.4ELISA检测肝匀浆IL-1β和TNF-α的表达 剪取-80 ℃冻存的肝组织按1 ∶9(g ∶ml)加入相应的PBS匀浆肝组织,留取上清液,严格按照试剂盒说明书操作步骤测定IL-1β、TNF-α。
1.3.5Western blot检测肝脏Btk、p-Btk、NF-κB、NLRP3蛋白表达 剪取适量的肝组织加入RIPA裂解液充分匀浆提取总蛋白。用BCA法进行蛋白定量。加入SDS-PAGE (5×) 蛋白上样缓冲液,沸水中煮沸 5 min,-80 ℃ 冰箱中冻存。以β-actin(1 ∶1 000)作为内参,以抗Btk抗体(1 ∶3 000)、抗p-Btk抗体(1 ∶1 000)、抗NF-κB抗体(1 ∶5 000)、抗NLRP3抗体(1 ∶2 500)和抗β-actin抗体(1 ∶1 000)作为一抗进行聚丙烯酰胺凝胶电泳。采用自动化学发光系统拍摄分析。
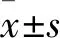
2 结果
2.1 ALT和AST酶活性检测结果与对照组比较,APAP腹腔注射0.5、1 h组小鼠血清ALT、AST水平差异无统计学意义(P>0.05);给药后3 h血清ALT、AST升高,持续至24 h仍维持在较高水平(P<0.01),36、48 h血清ALT、AST下降,但仍高于对照组(P<0.01)。见表1。
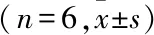
表1 不同时间点血清ALT、AST变化
2.2 肝组织病理检查结果对照组,肝小叶结构正常,未见肝细胞变性、坏死以及肝窦充血。APAP处理后0.5、1 h肝脏病理形态学与对照组比较差异无统计学意义。APAP处理后3 h肝窦充血,可见点状坏死灶,APAP处理后6、12 h肝窦充血明显,坏死面积逐渐增大,持续至24 h,主要表现为小叶中心性坏死;APAP处理36 h后肝脏病理变化逐渐减轻,坏死面积逐渐减少。APAP干预后的各组小鼠肝脏坏死程度较对照组有变化(F=29.845,P=0.000)。见图1、2。
2.3 肝组织匀浆IL-1β、TNF-α检测结果与对照组比较,APAP腹腔注射1、3 h后,TNF-α、IL-1β分别开始升高,且均在24 h时含量最高(P<0.01),后逐渐降低。见表2。
2.4 Western blot 检测结果与对照组比较,APAP干预后的各组小鼠肝脏Btk(F=441.280,P=0.000)、p-Btk(F=1 323.022,P=0.000)、NF-κB(F=133.125,P=0.000)及NLRP3水平(F=820.095,P=0.000)含量均有变化。正常肝组织中仅有少量的Btk表达,APAP处理组小鼠随着给药时间的延长,Btk及p-Btk的表达逐渐增多,于24 h表达最高(P<0.01),后表达逐渐下降。NF-κB、NLRP3表达与Btk及p-Btk表达变化趋势相一致。见图3。
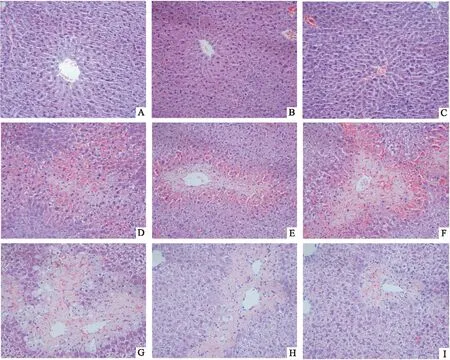
图1 不同时间点肝脏病理学变化 × 200
表2 不同时间点肝组织IL-1β、TNF-α表达量

图2 不同时间点小鼠肝脏坏死面积
3 讨论
APAP引起的肝损伤病理组织学主要表现为肝小叶中心性坏死[6-7]。ALT、AST是目前评价肝脏损伤程度的常用指标。本实验结果显示,小鼠单次腹腔注射APAP(300 mg/kg)0.5、1 h,血清ALT、AST及肝脏病理形态学与对照组无明显差异;其余各组血清中ALT、AST水平、3 h后各组肝细胞坏死面积明显高于对照组,同时肝脏HE染色也可见不同程度的肝窦充血。这些生化及病理改变与相关文献报道[6-7]一致,提示APAP诱导的小鼠肝损伤模型复制成功。本实验研究发现APAP(300 mg/kg)单次干预造成的肝损伤并非完全随时间的延长而加重,36 h后肝损伤的程度有所减轻,提示单次APAP造成的小鼠肝损伤存在自身修复过程。

图3 各组小鼠肝组织Btk、p-Btk、NF-κB、NLRP3蛋白表达情况
APAP诱导的肝细胞坏死的发生与过量服用APAP或肝脏基础水平还原型谷胱甘肽被耗竭,代谢产生的N-乙酰对苯醌亚胺不能被肝脏还原型谷胱甘肽结合,造成肝细胞代谢损伤和氧化应激损伤,破坏线粒体膜等有关[1]。Btk是一种非受体型酪氨酸激酶,表达于除T淋巴细胞及浆细胞的髓系细胞。Btk通过调控中性粒细胞募集和激活参与炎症过程[5]。当细胞受到细菌、真菌、病毒及其他危险信号的刺激时,Btk以磷酸化的形式活化后可直接调节NLRP3或间接调节NLRP3等信号进而引起炎性因子的释放[3-4]。Btk参与调控多个重要炎症信号转导通路中许多关键的信号蛋白,包括NF-κB、NFAT、PKC、Pin1、TLRs、caveolin-1 等[8]。近期研究[5]表明在小鼠肝脏缺血再灌注损伤模型中,Btk抑制剂可减轻缺血再灌注引起的肝脏炎性损伤。本实验结果显示:正常小鼠肝组织中仅有少量Btk表达;APAP作用24 h内,随着APAP作用时间的延长,小鼠肝组织中Btk及p-Btk逐渐升高,肝损伤的严重程度与Btk及p-Btk表达变化趋势相一致。提示Btk的活化参与了APAP肝损伤病理的形成。
Btk是先天免疫炎性反应中的关键因子,参与调控炎症信号转导通路中许多关键的信号蛋白[4,8],其参与APAP诱导的肝损伤可能作用机制为:① 激活NF-κB导致NLRP3形成及炎性因子升高。文献报道Btk/NF-κB信号通路可参与视神经脊髓炎的发病,引起急性炎症[9]。NF-κB可激活NLRP3炎性体造成炎性因子过度表达,进而导致炎症和自身免疫疾病的进展[10-11]。抑制Btk、NF-κB信号通路可显著控制炎症及减轻疾病[12-13]。通过药物或遗传手段抑制Btk可严重抑制NLRP3炎性小体的激活,减轻IL-1β、IL-6、TNF-α等炎性因子的释放,进而减轻缺血性脑损伤引起的炎性反应[14]。本实验结果显示,正常小鼠肝组织中仅有少量Btk表达,随着APAP作用时间的延长,小鼠肝组织中Btk及p-Btk逐渐升高,NF-κB、NLRP3、IL-1β、TNF-α表达也逐渐升高,肝损伤的严重程度与Btk及p-Btk、NF-κB、NLRP3、IL-1β、TNF-α表达变化趋势相一致。表明Btk磷酸化可能激活了NF-κB导致NLRP3形成及炎症因子升高引起了肝损伤。② 直接激活NLRP3炎性小体。NLRP3炎症小体的激活一方面主要通过激活NF-κB通路上调NLRP3和 pro-IL-1β的蛋白表达,另一方面线粒体DNA,穿孔毒素,病原体相关成分等信号也参与其激活[15]。近年来有文献[14]报道Btk是NLRP3炎性小体的重要组成部分,在炎症小体中起重要作用,可通过物理方式直接与NLRP3、ASC相互作用,激活炎性反应,引起炎性损伤。但APAP导致的急性肝损伤中,Btk激活NLRP3的确切机制尚有待进一步探讨。
综上所述,本研究结果显示,Btk在单次APAP致肝损伤的过程中存在动态变化,APAP诱导的肝损伤中Btk、p-Btk、NF-κB、NLRP3、IL-1β、TNF-α表达变化与肝损伤的严重程度基本相一致,表明Btk可能在APAP肝损伤中起着重要作用,将为寻找APAP肝损伤新的防治靶点提供了理论基础。
猜你喜欢
中西医结合心脑血管病杂志(2022年19期)2022-11-19
中国临床解剖学杂志(2022年1期)2022-11-15
中国现代医生(2022年21期)2022-08-22
检验医学与临床(2022年12期)2022-06-27
昆明医科大学学报(2021年12期)2021-12-30
临床肺科杂志(2021年4期)2021-12-23
现代临床医学(2021年4期)2021-07-31
北京大学学报(医学版)(2020年6期)2020-12-14
食管疾病(2020年4期)2020-12-08
分析化学(2018年12期)2018-01-22
