
Btk抑制剂依鲁替尼对高糖刺激巨噬细胞炎症的抑制作用及机制
2020-09-09 08:21许思敏吴永贵
安徽医科大学学报 2020年8期
许思敏,范 哲,吴永贵
糖尿病肾病(diabetic nephropathy, DN)是糖尿病重要的微血管并发症之一。研究[1]表明DN的发生发展与炎症有很大关系,其中肾脏组织巨噬细胞的浸润与肾脏组织炎症有着密切的联系。Bruton酪氨酸激酶(Bruton's tyrosine kinase, Btk)是一种非受体型酪氨酸激酶,研究[2]表明Btk不仅参与了获得性免疫调节,在固有免疫调节中也起到了重要作用,例如产生炎症因子单核细胞趋化蛋白1 (monocyte chemo-attractant protein-1, MCP-1),肿瘤坏死因子α (tumor necrosis factor-alpha, TNF-α),白细胞介素1β (interleukin-1beta, IL-1β)[3]。此外,研究[4]表明Btk活性可以通过与Toll样受体(如TLR2和TLR4),相互作用后产生,增强NF-κB p65亚基的激活作用。总之,Btk在炎症和免疫反应中发挥重要作用。
依鲁替尼(ibrutinib),又名PCI-32765,是一种高度选择性的小分子Btk抑制剂,通过共价结合半胱氨酸-481 (cysteine 481, Cys-481)抑制Btk活性[5]。PCI-32765近年来在临床前研究和临床试验中均显示出很强的抗淋巴瘤活性[6],此外,在动物自身免疫性疾病模型,如类风湿性关节炎中也有使用[7]。该研究主要探讨PCI-32765对高糖刺激巨噬细胞释放炎症因子的影响及其机制。
1 材料与方法
1.1 抗体与试剂葡萄糖、甘露醇购自美国Sigma公司;Btk抑制剂PCI-32765购自美国Selleck公司;试剂盒试剂购自美国Invitrogen公司;SYBR Green荧光定量PCR Master Mix试剂盒、Revert Aid第一链cDNA合成试剂盒、CCK-8试剂盒均购自南京诺唯赞生物科技有限公司;FITC标记的抗小鼠F4/80抗体、APC标记的抗小鼠/抗人CD11b抗体及同型对照均购自美国Bio Legend公司;Btk抗体、细胞外信号调节蛋白激酶(extracellular regulated protein kinases, ERK)抗体、c-Jun氨基末端激酶(c-Jun N-terminal kinase, JNK)抗体、p38丝裂原活化蛋白激酶(mitogen-activated protein kinase, MAPK)抗体、核因子κB(nuclear factor kappa B, NF-κB) p65抗体和核转录因子抑制蛋白(inhibitor of kappa B, IκB)抗体及其相应磷酸化产物p-Btk抗体、p-ERK抗体、p-JNK抗体、p-p38MAPK抗体、NF-κB p-p65抗体和p-IκB抗体以及诱导型一氧化氮合酶(inducible nitric oxide synthase, iNOS)抗体均购自美国Cell Signaling Technology公司;β-actin抗体、辣根过氧化物酶标记的抗兔IgG抗体和抗鼠IgG抗体购自武汉三鹰生物技术有限公司;蛋白质检测试剂盒购自江苏碧云天生物技术研究所;检测鼠IL-1β的ELISA试剂盒均购自广州瑞博奥生物科技有限公司;检测鼠MCP-1、TNF-α的ELISA试剂盒购自上海依科赛生物科技有限公司;ECL发光液购自美国Thermo Scientific公司。
1.2 骨髓来源巨噬细胞(bonemarrow-derivedmacrophages,BMMs)的培养BMMs由雄性C57小鼠分离获得。选用6~8周龄雄性C57小鼠的股骨和胫骨,去除肌肉及结缔组织,用75%乙醇浸泡消毒3 min后,将股骨和胫骨移至4 ℃预冷的无菌PBS溶液中漂洗1 min。在超净台剪断骨的两端,用1 ml注射器缓缓将骨髓腔内细胞冲洗进含2%的胎牛血清(fetal bovine serum, FBS) 4 ℃预冷的无菌PBS溶液(约6 ml/只)中。将含有骨髓腔内细胞的冲洗液经2 290 r/min 4 ℃离心10 min,弃去上清液,加入3 ml红细胞裂解液去除红细胞,2 290 r/min 4 ℃ 离心5 min。将沉淀的细胞重悬于含10 %FBS,1%青链霉素及15% L929细胞上清液的DMEM低糖培养基(5 mmol/L)中,在37 ℃,5% CO2的培养箱中培养3 d后更换培养基,此后每天更换。7 d后采用流式细胞术检测BMMs的成熟度及纯度。
1.3 细胞活力检测采用CCK-8试剂盒检测细胞活力。在96孔板每个孔中加入1×104个BMMs。24 h后,使用不同浓度的PCI-32765或二甲基亚砜处理BMMs 45 min,再予以高糖刺激24 h。向每个孔中加入10 μl CCK-8溶液,继续培养4 h。用酶标仪检测490 nm下各孔的吸光度(OD)值。细胞活力的计算:细胞活力=(加药细胞OD-空白OD)/(对照细胞OD-空白OD)×100%。将低糖组的平均细胞活力定为100%,其余实验结果以百分比表示。
1.4 实验条件的优化与实验分组采用20~35 mmol/L高糖刺激BMMs,使用甘露醇作为渗透压对照,选定影响Btk表达及BMMs活化的最佳浓度作为实验中的高糖浓度。采用10-9~10-5mmol/L PCI-32765影响高糖刺激BMMs,选定抑制Btk表达的最佳浓度作为实验中的抑制剂浓度。根据研究目的和实验条件优化结果,将BMMs分为低糖组(LG),抑制剂对照组(LG+PCI-32765),高糖组(HG)和抑制剂组(HG+PCI-32765),其中,LG组葡萄糖浓度为5 mmol/L。
1.5 RNA提取及qRT-PCRRNA提取和qRT-PCR的操作方法参考文献报道[8]。使用TRIzol试剂从实验所得BMMs中提取总RNA。通过逆转录反应体系将1 μg RNA逆转录为cDNA。采用SYBR Green荧光定量PCR Master Mix试剂盒完成实时定量PCR,实现cDNA扩增。检测mRNA的引物序列为包括:GAPDH作为内参:F: 5′-ACCCCAGCAAG GAGACACTGAGCAAG-3′, R: 5′-GGCCCCTCCTGTTA TTATGGGGGT-3′; TNF-α: F: 5′-CCCTCCTGGCCAA CGGCATG-3′, R: 5′-TCGGGGCAGCCTTGTCCCTT-3′; MCP-1: F: 5′-TTGACCCGTAAATCTGAAGCTAA T-3′, R: 5′-TCACAGTCCGAGTCACACTAGTTAC-3′; IL-1β: F:5′-GCCTCGTGCTGTCGGACCCATAT-3′, R: 5′-TC CTTTGAGGCCCAAGGCCACA-3′。使用2-ΔΔCt法计算mRNA相对GAPDH的表达量。
1.6 ELISA将BMMs根据实验要求按组别处理后从培养基中提取上清液,5 000 r/min 4 ℃离心10 min,离心后再次留取上清液。根据ELISA试剂盒说明书检测MCP-1、IL-1β、TNF-α。
1.7 激光共聚焦检测将细胞铺板于24孔板。在含有PCI-32765或不含PCI-32765的培养基中预处理45 min后,再加入高糖刺激24 h,室温下用4%多聚甲醛固定30 min。使用混合溶液(5%驴血清+0.2%聚乙二醇辛基苯基醚)封闭2 h后,加入iNOS或NF-κB p65一抗4 ℃孵育过夜。PBS洗涤3次后,加入F4/80标记的一抗Alexa Fluor 647和二抗Alexa Fluor 488,37 ℃避光孵育2 h。再次用PBS洗涤3次后,滴入DAPI染色细胞核10 min。使用Leica TCS SP5激光共聚焦显微镜(Leica公司,德国)观察巨噬细胞。
1.8 流式细胞术分析将成熟的BMMs按组别处理后分别收集细胞,每组1×106个,2 500 r/min离心5 min,弃去上清液,用PBS (500 μl/管)重悬细胞,加入CD16/CD32抗体,室温处理20 min后,2 500 r/min离心10 min,弃去上清液,再次以PBS(500 μl/管)重悬细胞,加入FITC标记的F4/80、APC标记的CD11b,于室温下避光孵育30 min。PBS洗涤两次后,加入500 μl PBS,用流式细胞术检测巨噬细胞。
1.9 Western blotBMMs在含有PCI-32765或不含PCI-32765的培养基中预处理45 min,再按实验设计予以高糖刺激。提取各组总蛋白并转移至PVDF膜上,以5%脱脂牛奶37 ℃封闭2 h后,加入一抗4 ℃孵育过夜。PBST洗涤3次后孵育相应二抗,孵育条件为37 ℃ 45 min。再次用PBST洗涤3次后,将PVDF膜置于ECL发光液中15~30 s后,采用化学发光凝胶成像系统检测蛋白表达情况。最后,使用ImageJ软件统计各条带灰度值,并与内参比较分析。
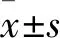
2 结果
2.1 BMMs的鉴定巨噬细胞采用FITC标记的F4/80抗体和APC标记的CD11b两种抗体标记。如图1A所示,F4/80及CD11b均阳性表达的细胞判定为成熟的BMMs。第7天获得纯度为94.5%的巨噬细胞。
2.2 实验条件的优化如图1B所示,Western blot分析结果显示在不同浓度的高糖组中,30 mmol/L的高糖组中p-Btk和iNOS表达较5 mmol/L低糖组增加最多,差异有统计学意义(F=108.938,P<0.05;F=79.779,P<0.05),因此采用30 mmol/L作为实验中高糖浓度。如图1C所示,PCI-32765在10-6mmol/L和10-5mmol/L对p-Btk表达抑制效果最佳(F=4 285.000,P<0.01;F=426.276,P<0.01),且两者之间的差异无统计学意义,故选择较低浓度10-6mmol/L作为实验中抑制剂对照组和抑制剂组中PCI-32765浓度。
2.3 PCI-32765对BMMs活力的影响为了确定PCI-32765是否影响BMMs活力,采用不同浓度的PCI-32765预处理巨噬细胞45 min,再加入高糖刺激24 h。如图1D所示,与未处理细胞相比,10-8~10-5mmol/L PCI-32765无明显细胞毒性,当PCI-32765浓度达到10-4mmol/L时,高糖刺激下的骨髓巨噬细胞活力受到影响,差异有统计学意义(F=34.287,P<0.05)。
2.4 PCI-32765对BMMs趋化的影响为了检测PCI-32765对BMMs趋化功能的影响,将高糖诱导的BMMs分为2组,一组使用PCI-32765处理,另一组不使用PCI-32765,分别检测细胞趋化情况。结果显示,高糖刺激的BMMs趋化量高于低糖组(F=191.919,P<0.05),而10-6mmol/L PCI-32765预处理后,BMMs趋化受到抑制(F=18.901,P<0.05) (图2)。

图1 BMMs的鉴定
2.5 PCI-32765对BMMs iNOS表达的影响为了确定PCI-32765是否能改变BMMs极化,采用共聚焦显微镜分析BMMs的表型。结果显示,未处理的巨噬细胞中绿色(iNOS)荧光较弱,而在高糖诱导的BMMs中,观察到更强的iNOS染色,经PCI-32765处理后iNOS染色减弱(图3A)。Western blot分析结果提示,高糖增加了iNOS的表达(F=173.206,P<0.01),PCI-32765降低了iNOS的表达(F=22.289,P<0.05)(图3B)。
2.6 PCI-32765对高糖刺激的BMMs炎症因子分泌和mRNA水平的影响用ELISA法分别检测各组培养基上清液中TNF-α、 IL-1β、 MCP-1含量。如图4A所示,高糖组中TNF-α、 IL-1β和MCP-1的水平高于低糖组,差异具有统计学意义(F=10.882,P<0.05;F=70.218,P<0.05;F=349.905,P<0.05)。而相比高糖组,PCI-32765抑制了高糖刺激产生的这些细胞炎症因子,差异有统计学意义(F=30.477,P<0.05;F=22.599,P<0.05;F=370.476,P<0.05)。用qRT-PCR法分别检测各组BMMs中TNF-α、IL-1β和MCP-1的mRNA相对表达量。如图4B所示,高糖组中,TNF-α、 IL-1β和MCP-1的mRNA水平高于低糖组,差异有统计学意义(F=80.279,P<0.05;F=15.987,P<0.05;F=505.917,P<0.05),而PCI-32765抑制了其mRNA的表达(F=285.553,P<0.05;F=321.007,P<0.05;F=1 588.000,P<0.05)。
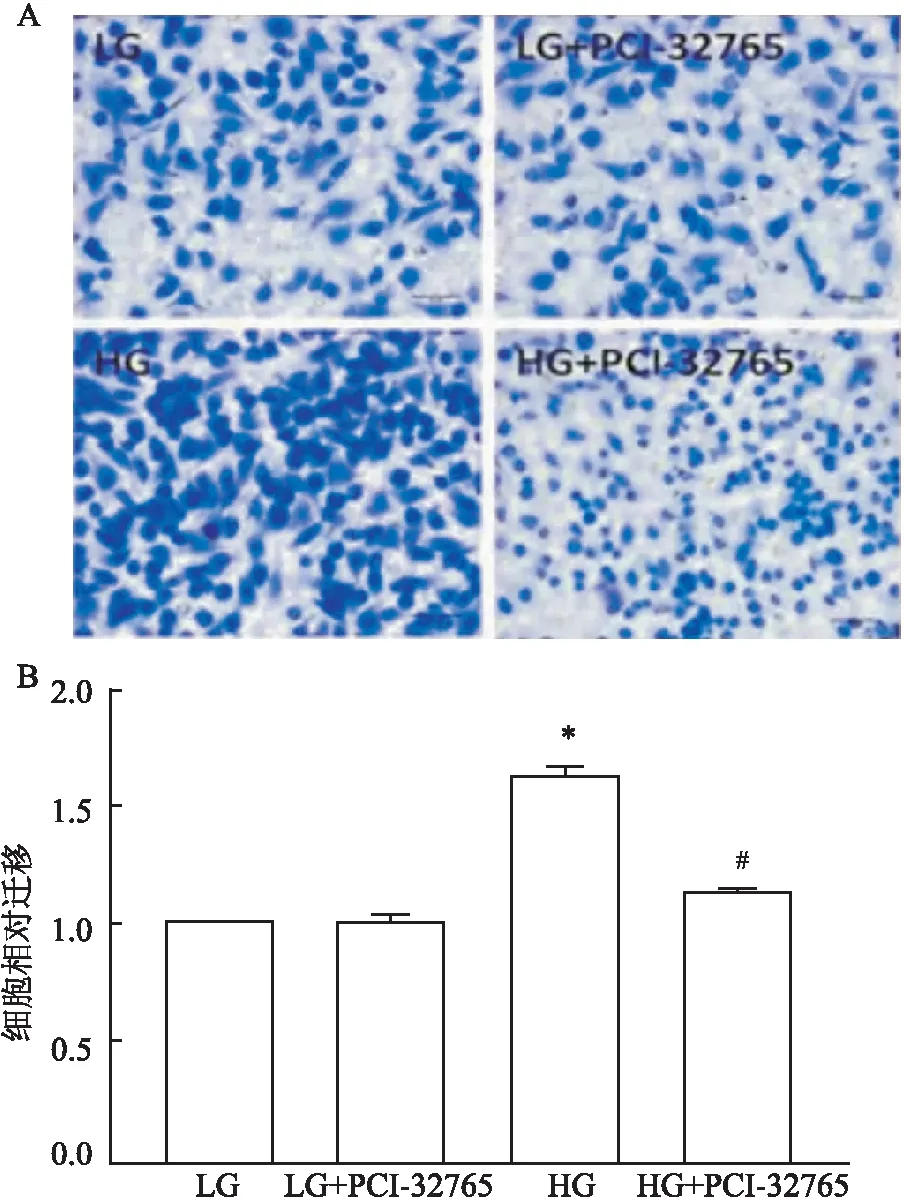
图2 PCI-32765对高糖刺激BMMs趋化作用的影响
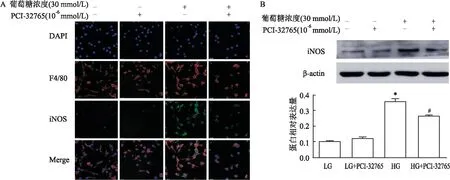
图3 PCI-32765对高糖刺激BMMs活化的影响

图4 PCI-32765对高糖刺激BMMs分泌促炎因子及mRNA表达的影响
2.7 PCI-32765对高糖刺激的BMMs中p-Btk、p-p38MAPK、p-ERK、p-JNK、NF-κB p-p65和p-IκB表达的影响检测高糖刺激下,不同时间MAPK及NF-κB信号通路各蛋白表达情况,根据最佳表达时间为选取高糖刺激时长。如图5、6所示,Western blot结果分析表明,高糖诱导的BMMs组中p-Btk表达增加(F=313.820,P<0.01),同时,p-p38MAPK、p-ERK、p-JNK和NF-κB p-p65水平亦有增加(F=3 824.000,P<0.05;F=1 083.000,P<0.05;F=6 147.000,P<0.05;F=2 833.000,P<0.05)。抑制剂对照组中上述蛋白表达相比低糖组无明显差别,PCI-32765抑制了高糖诱导的p-Btk、 p-ERK、NF-κBp-p65和p-IκB表达(F=154.631,P<0.05;F=8.654,P<0.05;F=6 546,P<0.05;F=12.91,P<0.05)(图7~9)。

图6 高糖刺激在不同时间点对BMMs各蛋白表达的影响
2.8 PCI-32765对p65核转移的影响为了研究PCI-32765在NF-κB信号路径中的作用,使用激光共聚焦检测p65核转移。研究结果表明,在低糖组中,绿色荧光标记的NF-κB p65几乎全部分布于细胞质;在高糖组中,绿色荧光主要分布在细胞核中。而PCI-32765抑制了高糖诱导的BMMs p65核转移。见图10。

图7 PCI-32765对p-Btk表达的影响

图8 PCI-32765对MAPK信号通路各蛋白表达的影响
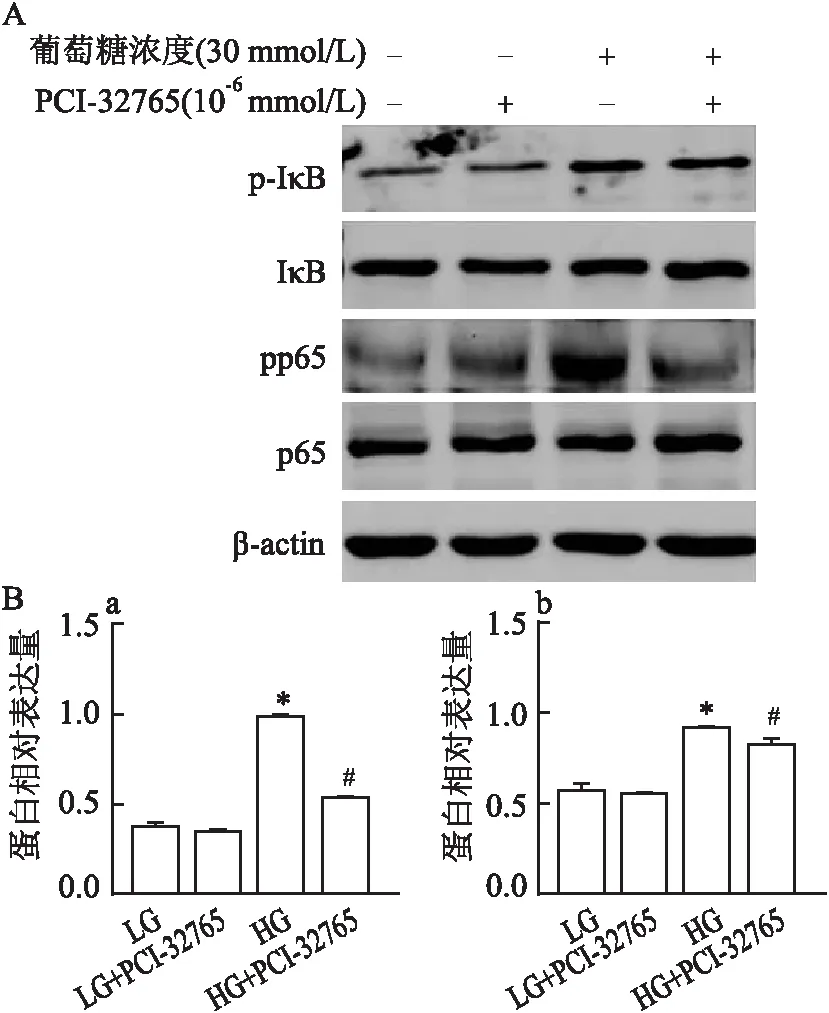
图9 PCI-32765对NF-κB信号通路蛋白表达的影响
3 讨论
在中国慢性肾脏病透析前人群中,DN已经超过肾小球肾炎成为慢性肾脏病的主要病因,在不久的将来,DN将不可避免地成为导致透析的主要原因[9],研究其致病机制有助于减少透析人群,减轻社会经济负担。DN主要病理改变为巨噬细胞聚集、肾小球肥大、基底膜增厚、细胞外基质增加以及肾脏纤维化,最终引起肾功能损害。目前对于DN的研究[10]提示巨噬细胞浸润程度与糖尿病的肾脏损害之间存在相关性。高糖导致巨噬细胞活化、增殖、分化为促炎性巨噬细胞,表达较高的iNOS,引起炎症反应和组织改变。
越来越多的学者研究DN与慢性微炎症的关系。本团队曾经报道[11]过,炎症介质如MCP-1和TNF-α,在糖尿病db/db小鼠的肾脏中表达增加。抑制炎性细胞因子的分泌可能延缓DN的进展。本研究结果表明,TNF-α、IL-1β和MCP-1的水平在高糖诱导的BMMs中增加,而高糖诱导的BMMs趋化较正常组织增加,提示高糖诱导巨噬细胞迁移的原因是上清中炎症介质浓度增加,而Btk抑制剂PCI-32765,可以抑制MCP-1、TNF-α及IL-1β这些细胞炎症因子的表达。这与高糖环境下iNOS表达激活巨噬细胞[12]、PCI-32765抑制巨噬细胞激活和炎症反应结果一致。

图10 PCI-32765对高糖刺激BMMs NF-κB p65核转移的影响
高糖引起ERK信号通路在系膜细胞和足细胞中激活[13-14]。本研究表明,高糖可以激活巨噬细胞中ERK、JNK、p38MAPK以及NF-κB信号通路,PCI-32765抑制了高糖诱导的ERK和NF-κB信号通路激活,而p-JNK和p-p38的减少并不明显,这进一步证实了Btk在ERK和NF-κB信号通路起到关键作用,同时在高糖诱导肾脏炎症中发挥了重要作用。其中,NF-κB通过调节细胞炎症因子的基因表达,参与肾脏炎症反应。Yang et al[15]报道,抑制NF-κB通路可以抑制高糖刺激足细胞产生的炎性细胞因子。本研究结果提示,抑制Btk可以降低高糖诱导BMMs中TNF-α、 IL-1β和MCP-1水平。因此,PCI-32765可能通过抑制NF-κB信号通路,从而在抗炎中发挥作用;这与研究[4]提示的Btk调节巨噬细胞NF-κB磷酸化的结论相一致。
本研究结果表明,高糖通过调节ERK、 JNK、 p38MAPK以及NF-κB信号通路,诱导巨噬细胞的炎症,参与DN发生。而Bruton酪氨酸激酶抑制剂PCI-32765,通过ERK及NF-κB信号通路降低了炎症因子TNF-α、IL-1β以及MCP-1水平,提示Btk可能参与高糖环境下巨噬细胞通过ERK及NF-κB信号通路激活,参与调节高糖诱导巨噬细胞炎症的发生,这为抑制DN炎症反应提供了新的研究方向。
猜你喜欢
日用电器(2022年7期)2022-09-07
眼科新进展(2022年2期)2022-03-11
中国果业信息(2021年7期)2021-12-01
现代临床医学(2021年5期)2021-11-02
天津医科大学学报(2021年4期)2021-08-21
天津医科大学学报(2021年4期)2021-08-21
现代临床医学(2021年2期)2021-03-29
凤凰生活(2019年8期)2019-08-16
中国新闻周刊(2016年32期)2016-10-27
食品安全导刊(2016年7期)2016-05-14
