
重大传染病疫情中临床试验质量的保障和对策探讨
——以新型冠状病毒肺炎为例
2020-06-28 07:59杜艾桦
医学与社会 2020年5期
杜艾桦 尹 平
1华中科技大学同济医学院附属同济医院科研处,武汉,430030;2华中科技大学同济医学院公共卫生学院,武汉,430030
新型冠状病毒肺炎(以下简称新冠肺炎)作为急性呼吸道传染病已纳入《中华人民共和国传染病防治法》规定的乙类传染病(国卫办医函〔2020〕184号),按甲类传染病管理。截至2020年3月22日24时,中国累计确诊病例81093例,其中湖北省67800例,武汉市50005例[1]。另据世界卫生组织(WHO)公布,海外截至欧洲中部时间3月22日10时共有186个国家和地区先后有新冠肺炎确诊病例报道,全球累计292 142例确诊病例[2]。为在检测试剂、有效药物和疫苗等方面取得重大突破,多家临床试验机构纷纷开展新冠肺炎相关临床研究。但是,面对如此多的患者,医护人员在前线为疫情奋战的同时还需要兼顾临床研究,临床质量如何得到保障成为当下关注的热点。本研究以疫情较为严重的武汉市开展的已获伦理批准的临床研究项目为分析对象,力图剖析目前临床研究存在的问题,为提高重大传染性疫情中的临床研究质量提供对策。
1 武汉地区新冠肺炎相关临床研究的现状
新冠肺炎疫情暴发以来,与疫情相关的临床研究出现井喷式增长,截至2020年3月22日,在“中国临床试验注册中心”(http://www.chictr.org.cn)网站上,“研究疾病名称”为“新型冠状病毒肺炎”或“COVID-19”检索项目出488项,附加“实施地点”省份为“湖北”的项目有246项,再追加“实施地点”市为“武汉”的项目有166项(其中已获伦理批准的有140项),武汉地区新冠肺炎临床研究项目约占全国的34%。如图1所示,武汉地区新冠肺炎新增确诊病例数在2月12号达到峰值,随后逐渐减少,而从1月下旬开始武汉地区新冠肺炎相关临床研究项目注册量一直在攀升,下文介绍的新冠肺炎相关临床研究注册情况均以“实施地点”为“武汉”且已获伦理批准的项目进行统计。
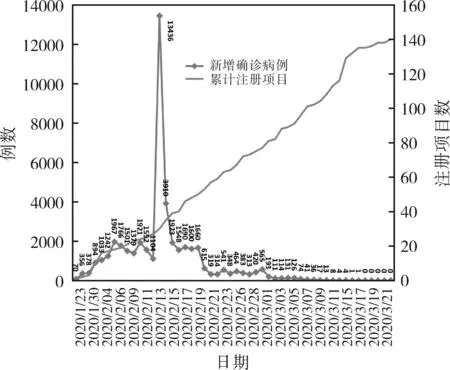
图1 截至2020年3月22日临床研究注册情况及每日新增确诊病例数
注:数据分别来自《中国临床试验注册中心》和武汉市卫生健康委员会网站“疫情通报”
在武汉地区开展的新冠肺炎相关项目申办单位都是各医疗机构,以研究者发起的临床研究为主,图2所示申办项目排名前十的医院,“华中科技大学同济医学院附属同济医院”“华中科技大学同济医学院附属协和医院”、“武汉大学人民医院”、“武汉大学中南医院”和“湖北省中医药”是通过认证的有十几年临床试验经验的老临床试验机构,其他几家机构都是最近几年才开始从事临床试验的新机构。
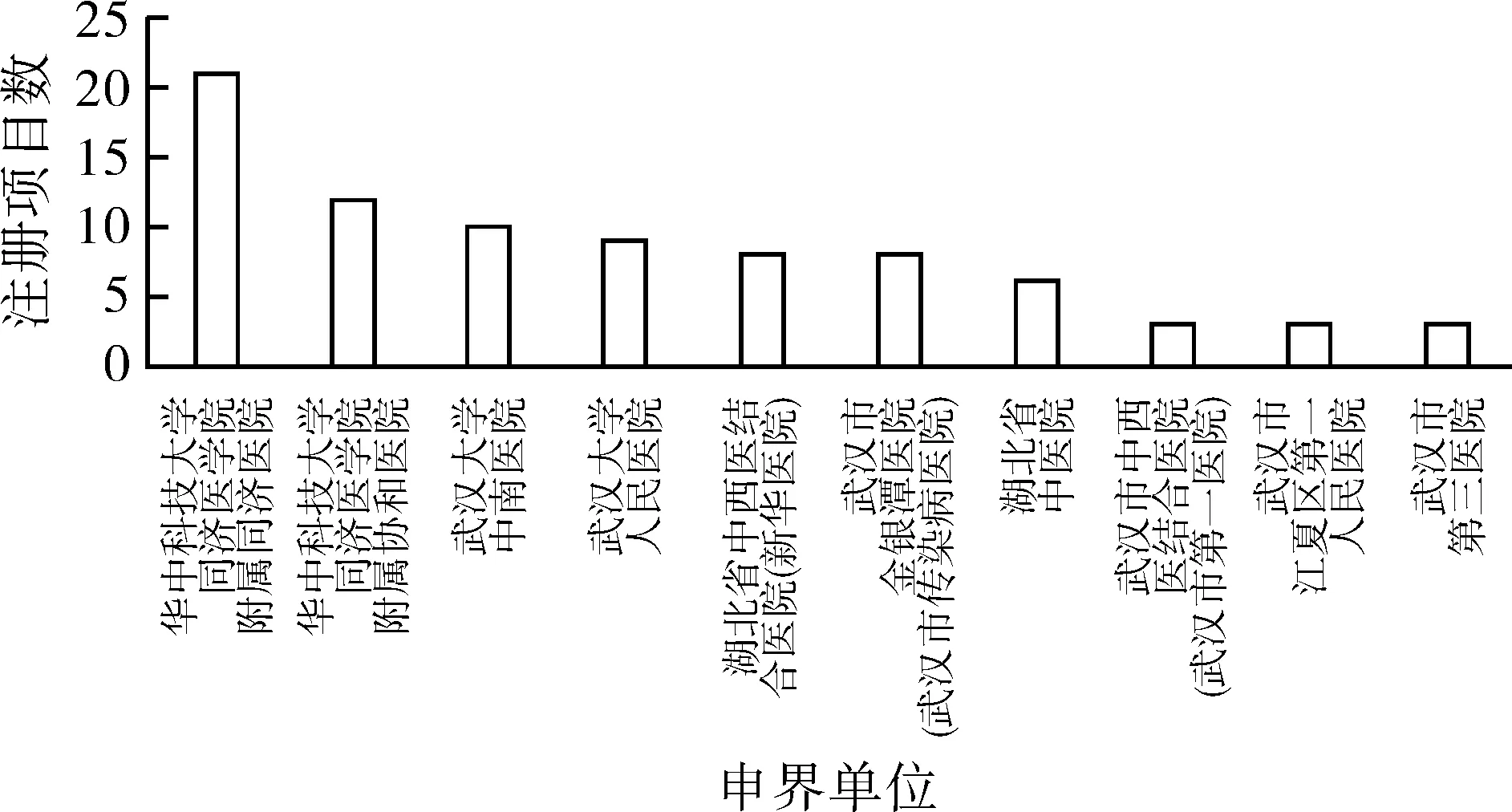
图2 新冠肺炎临床研究发起单位排名前十的申办单位情况
另外,在武汉地区实施的这140个项目,以干预性研究为主(75个),其次是观察性研究(53个)。如图3所示,在75个干预性研究中,又以中医中药相关项目最多,其次是免疫及生物制剂项目和化学药物项目,其中有很多项目研究相似或者重复(见表1),因为中西医结合(或中医)类项目有些注册信息只显示使用了中药,但是并未写明用的什么中药,故未做统计;另外,这75个干预性研究中,各项目受试者样本量分布情况如图4所示,相当多项目的受试者样本量范围为50≤样本量≤100,各项目的研究设计方法以“随机平行对照”为主,并且这些随机试验中采用双盲的仅有4项,单盲1项,其他都是开放性的研究(见图5)。
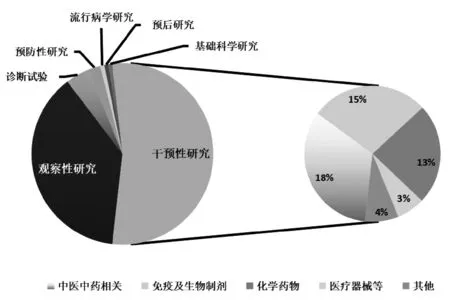
图3 项目研究类型组成情况
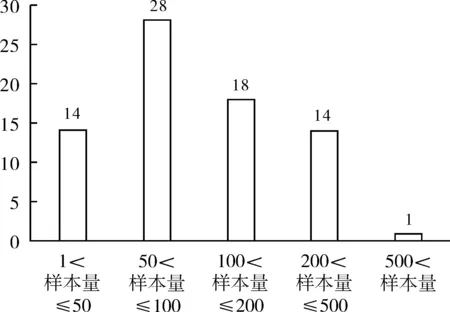
图4 干预性研究项目样本量分布情况
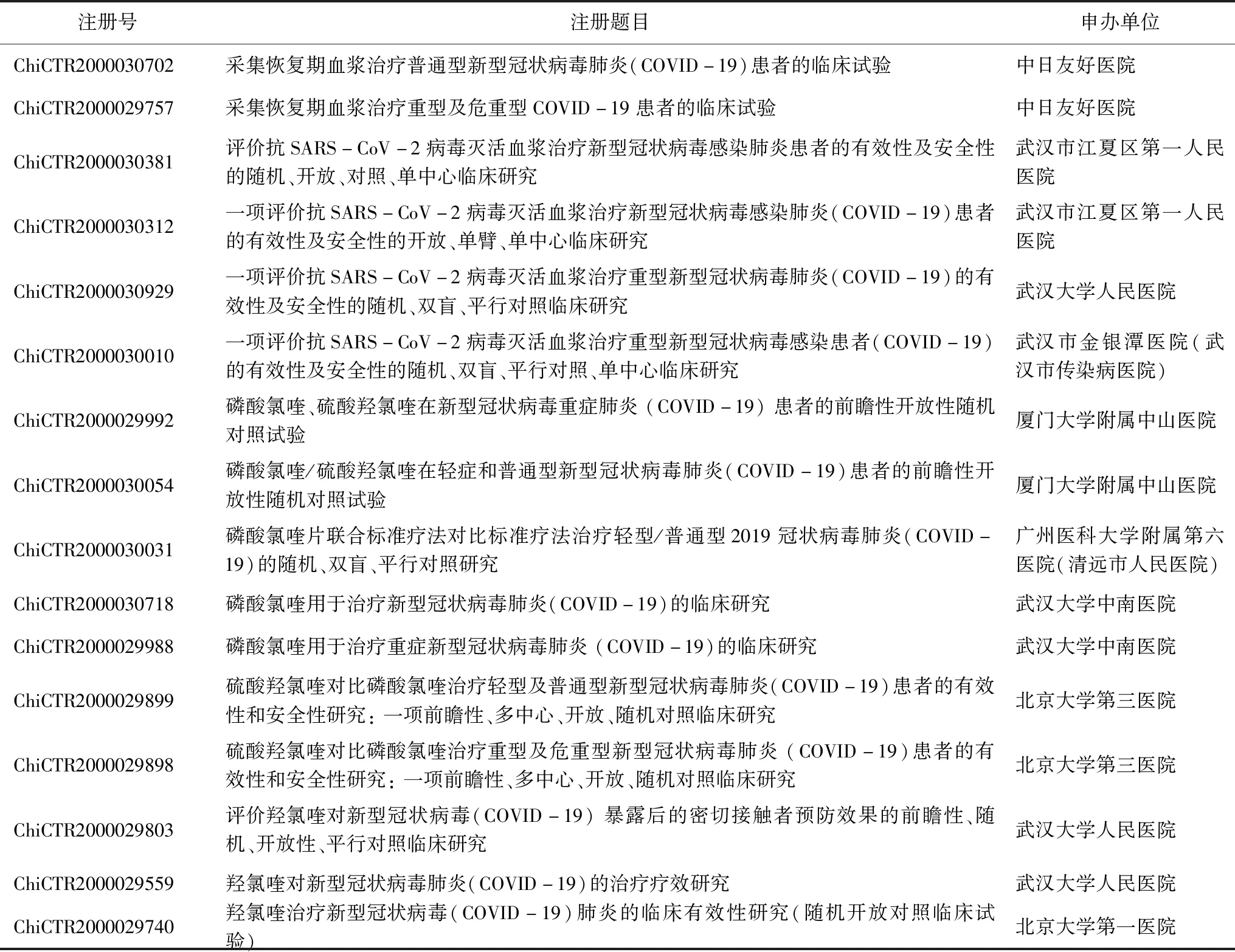
表1 干预性研究中研究对象相似的项目

图5 干预性研究项目研究设计方法
2 新冠肺炎临床试验中存在的问题
2.1 项目立项管理
从表1可见不少不同机构开展有相似或重复的研究,比如干预性研究中仅“磷酸氯喹”和/或“羟氯喹”项目有10个,“恢复性期血浆治疗”项目有6个。单个临床试验机构在项目立项审查时,应考虑优先次重、轻重缓急,积极支持最有希望对控制疫情有帮助的项目,避免一窝蜂涌向某一药物或研究某一问题,应尽量减少重复研究。但是,不同的研究机构还会存在重复研究问题,这一现象在疫情中也很凸显,也难避免。尽管这些相似的项目并不完全相同,但是在疫情严重和资源紧张的情况下是否有必要开展如此多的类似研究让人忧虑。WHO《传染病暴发伦理问题管理指南》[3]中强调“国家有关当局和国际组织应设法协调研究项目,以便确定与更广泛的疫情应对工作相一致的优先事项,并避免不必要的重复研究工作或不同研究中心之间的竞争”、“如果研究对正在进行的应对工作很重要,研究人员有义务共享所收集的信息作为研究的一部分”,并强调“确保研究不会耗尽与健康相关的重要资源”、“如果研究会过度占用其他重要的临床和公共卫生资源,包括人员、设备和保健设施,则不应进行研究。”这些规范性要求提示面对严峻的疫情和有限的资源,项目的立项管理应统筹安排。
2.2 临床试验设计的合理性问题
武汉地区140个已获伦理批准的项目中,多数采用了随机平行对照设计,但采用盲法,特别是双盲设计的研究不多。如果不采用盲法无疑会在涉及诸多受试者临床症状的评判中产生主观偏倚而影响研究结果。针对新冠肺炎而言,由于没有公认有效的治疗药物,“金标准”应该是在常规治疗的基础上使用安慰剂对照,进行优效试验设计。但这140个项目中,安慰剂对照不到10项。有不少试验是以常规治疗或所谓的阳性药物作为对照,而常规治疗繁杂,包括二十多种中成药、方剂以及西药抗病毒、抗菌、支持治疗等等,混杂效应大。在有效性评价指标方面,多集中在基于体征症状改善的病情缓解率、痊愈率,轻症或普通型向重症/危重症的转化率、病死率等方面。有些试验的随机化方法不明确或不恰当,各项目的样本量差距较大,存在很大的随意性。比如同是“干预性研究”、均为“随机平行对照”、研究对象都是“新冠肺炎轻症患者”的项目,“洛匹那韦/利托那韦”(注册号ChiCTR2000029539)分2个组,每组需要受试者164例,共计328例,而“甘草酸二铵肠溶胶囊联合维生素C片”(注册号:ChiCTR2000029768 )分2个组,每组需要受试者30例,共计60例。所有这些无疑都会影响试验药物有效性与安全性的客观、准确的评价,不能得出确切的结论。
2.3 受试者样本量问题
临床试验的样本量与试验的设计类型(非劣效、优效)、主要疗效指标及Ⅰ类错误水平α、检验效能power等因素有关,是试验结果可靠性的重要保证。图4展现75个干预性研究样本量的分布情况,样本量小,试验容易完成,所需要的精力、经费也少,但是研究结果的可靠性也较差,容易出现假阴性的结论;而样本量大,虽然试验结果可靠性高,但试验规模的增大,不仅增加了试验成本,更重要的是增加了完成试验的难度,特别是针对新冠肺炎这类爆发性的传染病,受试者入组的窗口期有限,要在比较短的时间内完成大样本的受试者入组并确保不违背试验的“入排标准”是有相当大的难度的。比如一个需要300例受试者的项目“ChiCTR2000030255”,有着较为严格的“入排标准”。纳入标准是:①符合《新型冠状病毒肺炎诊疗方案(试行第六版)》诊断标准,临床分型为普通型、重型;②中医辨证属风热证者;③年龄18-75周岁,性别不限;④知情同意,自愿受试并签署知情同意书。
其排除标准是:①原发性免疫缺陷病、获得性免疫缺陷综合征、先天性呼吸道畸形、先天性心脏病、胃食管反流症、肺发育异常等基础疾病引起的呼吸道感染,有明确细菌感染证据;②有以下状况的受试者:需每日治疗的哮喘,任何其他慢性呼吸道疾病,呼吸系统细菌感染如化脓性扁桃体炎,急性气管支气管炎,鼻窦炎,中耳炎等其他影响临床试验评估的呼吸道疾病。胸部CT证实存在严重的肺间质病变、支气管扩张等基础性肺部疾病患者;③重症肺炎需要机械通气者;④经研究者判断,既往或现在患有的疾病,可能影响患者参加试验或影响研究的转归,包括:恶性病、自身免疫性疾病、肝肾疾患、血液病、神经系统疾病、和内分泌疾病;现患有严重影响免疫系统的疾病,如:人类免疫缺陷病毒(HIV)感染,或血液系统,或脾切除、器官移植术等;⑤孕妇或哺乳期女性;⑥近3个月内参与过其他临床试验的患者;⑦过敏体质,如对两种或以上药物或食物过敏史者,或已知对本药成分过敏者;⑧研究者认为存在任何不适合入组或者影响受试者疗效评价的因素。仅仅排除标准⑥就能限制很多受试者的入组,通俗的讲就是一个新冠肺炎患者仅且仅能参加一项临床试验,有些项目的“入排标准”就将“近3个月”改成了“试验前28天”(ChiCTR2000031139 ),甚至直接删除该项要求(例如ChiCTR2000030288、ChiCTR2000030254、ChiCTR2000029988等)。如果放宽“入排标准”又会让样本人群中包含较多的混杂因素,不能保证试验的质量,影响研究结果[4]。
以华中科技大学同济医学院附属同济医院为例,目前,已经有60多项新冠肺炎相关临床试验项目与通过伦理审查,在《中国临床试验注册中心》和“clinical trials”临床试验注册网站却仅能查询到二十多项,还有很多项目因为各种原因尚未注册。其他临床试验机构应该存在类似情况,实际将要开展的项目数远比注册网站能查到的还要多。
截至2020年3月22日24时,湖北省现有新冠肺炎住院病例4593例,其中重症1343例[5],他们都是新冠肺炎研究的潜在受试者。
面对几百项的各类临床试验,确诊病例因治愈出院或死亡等原因在急剧减少,新增确诊患者也在逐步锐减(见图1),受试者样本量缺乏问题已越来越突出。且《新型冠状病毒肺炎诊疗方案》已经更新至第7版,推荐使用药物不断增加,目前住院患者病程长短不一,既往使用药物复杂,受制于临床试验严格的入排标准,多数临床试验特别是“入排标准”较严格的随机对照试验(RCTs)要筛选到合适的受试者会越来越难,甚至因受试者不足而不得不提前结束试验而无果而终。目前无序而缺乏统一协调的临床试验井喷式增长也无疑不利于重要药物的筛选而用于疫情控制。
2.4 研究者水平参差不齐
此次开展的新冠肺炎相关项目以研究者发起的临床研究项目为主,武汉地区开展新冠肺炎项目数排名前10位的医院中有一半是新成立的临床试验机构,承担项目较少,甚至有机构在新冠肺炎疫情前没有承担过临床试验项目,研究经验较少。即使承担项目较多的机构既往研究经验丰富,在疫情中研究团队协作也存在问题。由于疫情严重,病患较多,医护人员需求大,目前在一线参与救治的医护人员是从各个科室甚至不同的医院抽调组成的,以华中科技大学同济医学院附属同济医院为例,共有35支医疗志愿队,这些医护人员来自不同的医院,不管是支援医疗队还是本院医护在进行医疗救治的同时还需开展临床试验,如何快速高效的协作,如何严格执行研究方案是一个无法回避的现实问题。
2.5 伦理项目审查质量
疫情期间,伦理项目审查面临各种问题:①伦理审查的形式发生了改变,伦理审查会议不能像平常那样正常召开,会议只能以网络远程会议的模式开展,各伦理委员的工作状态不同、能力发挥受影响因素多,可能造成质量不佳的研究项目通过审查;②部分项目负责人在一线忙于救治,不能参加答辩,替代答辩的人对项目了解程度的不同影响答辩效果;③公共卫生当局也可能给予一定的压力,从而削弱伦理审查者的独立性;④由于疫情的紧迫性,短时间内申报项目数量多,项目审查要在短时间内给出意见,审查可能存在疏漏之处。
2.6 其他问题
自中国加入国际人用药品注册技术协调会(ICH)以来,药物和医疗器械临床试验的水平及规范性程度得到了极大的提高,临床试验分工细化和专业化,有专业的研究团队覆盖临床试验的各个环节,为临床试验的高质量提供了保证。而新冠肺炎疫情爆发以后,其他临床试验从业者,包括临床监察员(CRA)、临床协调员(CRC)、数据管理与统计分析人员等,因为封城、封路等原因不能到岗或因为职业暴露风险(未受过专业的医院感染等培训)尽量减少到医院开展临床试验相关工作。特别是疫情期间研究者发起的研究项目组建完善的研究团队都比较困难,再加上工作防护带来的困难,在如此仓促的条件下开展临床试验,缺乏其他专业的临床试验从业人员的协助,临床试验质量会受到一定的影响。
疫情期间,临床研究药物如何管理也是一个难题,很多临床研究的研究药物属于老药新用,医生可以开医嘱给患者调取使用,但一个病区患者由几组医护人员轮班管理,如何协调管理受试者的用药、临床研究用药物由专门的药品管理员管理还是药房统一管理、如何避免受试者合并用药等都是疫情下临床研究值得妥善解决的问题。
资料管理同样存在诸多问题,新冠肺炎病毒可通过接触传播,所以应尽量减少纸质资料,充分运用电子档案资料,如何管理电子资料、如何签署知情同意、如何收集填写研究数据、如何保障电子数据的安全性和保密性等都是需作相应准备;如果用纸质资料,纸质资料的发放回收以及消毒处理都值得重视。
3 对策
3.1 相关管理部门统筹协调临床试验项目
疫情期间,医护人员首要任务还是集中精力救治病人。国家和地方的相关管理部门应紧急成立管理团队,出台切合实际可操作的管理规定,有组织、科学地统筹协调临床研究项目,对项目的立项、实施、质量控制做出具体要求和安排,从源头做出科学有序的考虑。各医疗机构应按轻重缓急组建不同的攻关小组,对要解决的问题和有潜力的药物分门别类,组建不同的研究小组集中精力解决不同的问题。对理论上最有希望控制疫情的临床试验(如:瑞德西韦)应尽量避免一窝蜂大量上低质量的项目。未来疫情结束以后,医院要建立健全临床研究的管理制度,还应重视研究人员临床试验质量管理规范(GCP)和伦理知识的培训,提高研究者研究水平,这样才能有利于临床试验质量的提升。
3.2 加强区域伦理委员会的建设和审查
各地临床试验机构的伦理审查水平事实上存在差异,因此,特殊时期应该启用区域伦理委员会进行项目的综合审查,区域伦理委员会具有很多优势:委员专业背景强、审查质量高、审查效率高,有利统筹协调同一个地区的临床研究项目,提高审查质量,避免重复研究[6],减少受试者资源的浪费。疫情形势虽然严峻,伦理审查依然不能放松,伦理审查要把保护受试者的利益放在首位,方案设计中研究方法尽量不选择干预性研究方法,如果必须采用干预性研究,试验组和对照组均应保证受试者的基本治疗,在项目通过伦理审查后,更应做好实施过程中的跟踪审查[7]。
3.3 高度重视研究质量
研究质量是临床研究的生命,质量保证(QA)工作对于提高临床研究的质量至关重要,即使在重大传染性疫情期间也不能放松质量保证。在疫情期间,机构办可以培养自己的一个QA团队(外聘或者从内部培养),有完整的管理制度(SOP),有不同职责和分工,所有成员在熟悉GCP的同时,也掌握医院感染相关知识。该团队独立于各个研究项目组,负责对本机构各个项目进行质量控制,保证试验的有序进行和数据的产生、记录等都符合GCP要求[8]。作为中医药大国,我们有必要发挥中医中药的优势,但是目前中医药类临床研究各个环节还比较薄弱,QA团队可以帮助研究者在试验方案设计、选取操作性强的观察指标和临床试验过程等方面给予大力支持,让中医药类临床研究更科学的开展。
3.4 远程信息平台的建设
信息化系统使用是未来临床试验发展的趋势,数据在移动终端采集,在保证HIS、LIS等院内信息系统安全的前提下达成数据共享,简化手工录入等操作流程,电子数据逐步替代纸质数据,提高数据利用便捷度的同时有利于重大传染性疫情中数据的搜集和管理。数据传到信息平台后,很多非医护的临床试验从业人员可以通过平台,参与到临床试验中,将有助于数据管理和分析,提高临床研究数据管理质量。
4 结论
新冠肺炎疫情期间,为在检测试剂、有效药物和疫苗等方面取得重大突破,为尽快战胜疫情增添利器,各个临床试验机构都在积极开展相关临床研究。临床试验的质量就是临床试验的灵魂,直接关系到研究结果的可靠性。我们通过机构立项统筹安排、区域伦理委员会、机构QA体系和信息化平台建设等方面提出相关思考和对策,以期促进重大疫情下临床研究科学、有序的发展,全方位提高临床试验质量,产出高质量的成果服务于疫情中病人的救治,并最终控制疫情。
猜你喜欢
中国心血管杂志(2022年2期)2022-11-25
中国心血管杂志(2022年4期)2022-11-25
内蒙古统计(2021年4期)2021-12-06
中国心血管杂志(2021年6期)2021-01-02
基层中医药(2020年5期)2020-09-11
中国卫生统计(2019年3期)2019-07-10
中国心血管杂志(2019年3期)2019-01-04
中国卫生统计(2012年1期)2012-12-04
统计与决策(2012年14期)2012-07-25
中国合理用药探索(2012年2期)2012-03-20
