
着色性干皮病两例ERCC2和ERCC5基因突变分析
2020-06-05 06:53杨舟徐哲焦磊王珊马琳
中华皮肤科杂志 2020年4期
杨舟 徐哲 焦磊 王珊 马琳
国家儿童医学中心 首都医科大学附属北京儿童医院皮肤科100045
通信作者:马琳,Email:bch_maleen@aliyun.com
着色性干皮病(xeroderma pigmentosum,XP)是一组罕见的常染色体隐性遗传病,其发病机制为部分基因突变导致DNA 损伤修复能力受损,从而引发光敏、皮肤损伤、眼部病变、神经损害、恶性肿瘤、发育障碍等。皮肤癌为XP 患者的主要死因,神经退行性疾病是第二大死因[1]。目前报道XP 共有7个互补型(XPA、XPB、XPC、XPD、XPE、XPF、XPG)和1 个变异型(XPV),分别对应8 种基因(XPA、ERCC3、XPC、ERCC2、DDB2、ERCC4、ERCC5、POLH)突变[2]。本研究采用高通量测序对2 例XP患者进行全外显子组测序(whole ⁃ exome sequencing,WES),结合Sanger测序验证,明确其分别为XPD型及XPG型。
资料与方法
一、病历资料
例1 男,3 岁,出生时皮肤外观正常,日晒后面部出现红斑和肿胀,随后皮肤粗糙、干燥,逐渐出现散在的雀斑样病变和色素痣。皮疹夏季加重,以光暴露部位为主。近1年出现步态不稳,行走时易跌倒。体检:面、耳、颈部和双手背可见大量褐色斑点、斑片,间杂色素减退斑,散在疣状丘疹,躯干散发黑褐色斑丘疹(图1)。神经系统检查:步态不稳,易跌倒,四肢肌张力正常,深部腱反射可引出;指鼻试验、闭目难立征、轮替试验均阴性,未见共济失调;语言发育落后,无法表达完整的句子,智力筛查落后于正常同龄儿范围。其余系统检查未见明显异常。眼科检查正常;纯音测听未发现感音性耳聋;头颅磁共振成像检查大致正常,未见脑萎缩、脑室扩张、基底节及大脑皮质钙化等。
例2 男,1 岁5 个月,生后3 个月日晒后面部出现轻度潮红肿胀,后逐渐出现面部褐色斑点。体检:面部散在褐色斑点、斑片,伴少许色素减退性斑(图2)。体检:神经及其他系统未见异常。眼科、听力检查正常。
2例患儿均为第1胎第1产,父母非近亲结婚,无类似疾病家族史。嘱患者防晒、避光,随访2年,2例患儿皮损均较前有所增多,未出现恶性肿瘤。
二、方法
1.标本采集:用乙二胺四乙酸二钾抗凝管收集患儿及其父母外周静脉血3 ml,使用北京天根生化科技有限公司DNA 提取试剂盒提取DNA(总量>10 μg,50 mg/L)。本研究符合2013年修订的《赫尔辛基宣言》(https://www.wma.net/policies⁃post/wma⁃declaration⁃of⁃helsinki⁃ethical⁃principles⁃for⁃medical⁃research⁃involving⁃human⁃subjects/)的要求,患儿监护人及健康对照同意基因检测并签署知情同意书。
2.基因突变检测:①WES测序,稀释DNA后采用Covaris 超声仪随机分解成小片段DNA(180 ~280 bp),后经探针杂交和外显子区序列捕获,制备文库并质检,在北京康旭医学检验所应用美国Illumina 公司的Hiseq X Ten 测序平台进行测序,应用BWA(v0.7.15)软件将测序read与人类参考基因组对比,应用GATK(v3.6)软件检测单核苷酸变异和插入/缺失突变;②注释分析,使用ANNOVAR 注释软件注释变异在基因和转录本中的位置,基因相关注释涉及RefSeq、Ensembl、UCSC等数据库,利用千人基因组、GnomAD、TOPMED 等数据库注释变异在人群中的频率,使用蛋白损伤分析工具PolyPhen⁃2、SIFT 及Mutation Taster 进行致病性分析,利用OMIM、HGMD、ClinVar 等数据库对疾病进行注释,重点关注着色性干皮病相关基因XPA、ERCC3、XPC、ERCC2、DDB2、ERCC4、ERCC5 及POLH;③Sanger测序验证,针对单基因所需要验证的位点所在外显子/内含子的上下游序列设计引物(表1),对患儿突变位点进行PCR扩增,反应条件:95 ℃预变性10 min,95 ℃变性30 s、60 ℃复性30 s、72 ℃延伸45 s、共35个循环,72 ℃延伸5 min。PCR产物纯化后用ABI 3 730测序仪进行Sanger双向测序,获得数据后对结果进行判读,与Ensembl genome browse 90公布的序列比对分析。进一步对患儿父母进行验证,确定突变来源。
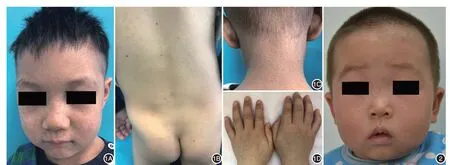
图1 例1临床表现 面部(1A)、背部(1B)、颈部(1C)、双手背(1D)散发大量褐色斑点、斑片,伴色素减退斑,散发疣状丘疹 图2 例2面部散发褐色斑点、斑片,伴少许色素减退斑
结 果
例1存在ERCC2基因复合杂合突变,即父源突变c.1805G>A(p.Gly602Asp)和母源突变c.586C>T(p.Arg196Ter)(图3),患儿符合XPD 型。例2 存在ERCC5 基因复合杂合突变,即父源突变c.2533+2T>C 和母源突变c.2453C>T(p.Ala818Val)(图4),患儿符合XPG 型。例1 的ERCC2 基因突变c.586C>T导致编码第196号氨基酸Arg的密码子变成终止密码子,为无义突变;例2 的ERCC5 基因突变c.2533+2T>C为编码区第2 533位核苷酸后11位内含子中第2位核苷酸由T变为C,为剪切变异,经fruitfly 网站(www.fruitfly.org/seq_tools)剪切位点预测工具预测,突变将导致原有的剪切位点散失,很可能无法正常剪切,导致蛋白功能出现异常。以上两个突变既往未见报道,在千人基因组、GnomAD、TOPMED 等健康人群对照数据库中未发现或频率极低,不属于多态性位点。而ERCC2 基因突变c.1805G>A 和ERCC5 基因突变c.2453C>T 为已报道的致病性变异,使用SIFT、Polyphen⁃2和MutationTaster 软件对2 个错义突变c.1805G>A 和c.2453C>T 进行功能预测,均分别为“有害”、“很可能损害”和“致病性”,显示其可能致病。Mutation Taster 软件对无义突变c.586C>T 预测为“致病性”,可产生截短蛋白,影响蛋白功能。PolyPhen⁃2软件显示分析c.1805G>A、c.2453C>T 及c.586C>T 突变所在位点均在不同物种间高度保守。

表1 ERCC2和ERCC5基因Sanger测序引物序列

图3 例1着色性干皮病患儿ERCC2基因复合杂合突变测序图 3A:先证者发生错义突变c.1805G>A(p.Gly602Asp)和无义突变c.586C>T(p.Arg196Ter);3B:先证者父亲发生错义突变c.1805G>A;3C:先证者母亲发生c.586C>T无义突变 图4 例2着色性干皮病患儿ERCC5基因复合杂合突变测序图 4A:先证者发生(父源)剪切突变c.2533+2T>C和(母源)错义突变c.2453C>T(p.Ala818Val);4B:先证者父亲发生剪切突变c.2533+2T>C;4C:先证者母亲发生错义突变c.2453C>T
讨 论
XPA ~XPG型XP是由涉及核苷酸切除修复机制的蛋白基因缺陷引起。NER可修复紫外线、化学试剂、毒性物质及化疗药物等引起的DNA损伤,以及一些因氧化还原过程产生的损伤[3]。不同分型的XP患者临床表现有所差异,不同地域种族XP分型亦有差异。在日本XPA 占较大比例;在美国XPC 占优势,其次为XPD 或XPV;在欧洲XPC 占主要比例,其次为XPV[4]。检索HGMD Pro、PubMed及万方和CNKI 数据库显示,截至2019 年10 月,我国XPA、XPC占优势[4⁃7],XPV并不少见[5,7⁃8],而XPD仅报道2 例[5⁃6],XPG 报道5 例[5⁃7]。本文报道XPD、XPG各1例,其中XPD为我国少见类型。与其他地区人群XPG罕见相比,我国XPG并不少见,且本例患者症状较轻。
XPD(OMIM#278730)由ERCC2 基因突变引起5′⁃3′ATP 依 赖 的DNA 解 链 酶XPD 活 性 降 低 所致[9]。目前已报道100 余种与疾病相关的ERCC2基因突变,其基因型与表型关系复杂,可导致多种症状且严重程度差异很大,如XPD、XPD 合并Cockayne综合征、毛发硫营养障碍、XP合并毛发硫营养障碍或脑-眼-面-骨骼综合征[4,10]。XPD患者常早期发病,临床表现严重(但轻于XPA),伴急性晒伤反应,雀斑样色素沉着明显,皮肤癌发生率较高[10]。XPD 患者多数可出现原发性神经元变性导致的进行性神经系统损害,如进行性步态不稳、肌张力低下、痉挛、癫痫等。这些症状开始于生命早期,亦可较晚出现,特征为髓鞘形成不足或异常、脑萎缩、钙沉积和脑细胞异常[3]。患者神经系统症状不能完全用紫外线损伤修复缺陷解释,还可能涉及核苷酸切除修复蛋白参与活性氧产生的损伤修复[3]。
我国既往仅报道2例XPD患者,均为复合杂合错义突变所致,尚未发现神经系统症状,其中p.Gly47Arg 位于HD1 区,p.Ile595Ser、p.Gly602Asp、p.Arg666Trp 均位于HD2 区(图5A)[5⁃6]。位于HD2区的突变可导致XP、XP 合并Cockayne 综合征、毛发硫营养障碍或XP 合并毛发硫营养障碍表型[4]。本研究中例1 患儿存在错义突变和无义突变的复合杂合突变,其错义突变p.Gly602Asp 位于HD2 区域(图5A),既往报道可引起XP合并Cockayne综合征。另有研究者报道HD2 区中突变Arg683Trp 可导致XP表型及神经系统疾病进展[4],而Arg683Gln可导致神经系统轻度异常或晚期发作[10]。p.Arg196Ter 为无义突变,产生截短蛋白,且位于4FeS 簇区域(图5A),推测其对酶活性影响较大。位于4FeS 簇区的突变可导致XP 或毛发硫营养障碍表型,其中I199Pfs*52 突变亦可导致XP 表型[11]。本文例1目前已出现大量雀斑样色素沉着、皮肤异色、疣状丘疹的XP皮肤表现,以及神经系统异常如步态不稳、语言及智力发育落后。有研究显示,合并神经退行性疾病的XP 患者更早发生皮肤癌[2],故该患儿需严格避光防晒,预防皮肤肿瘤发生。XP 患者发生脑肿瘤的风险可增加50 倍,包括髓母细胞瘤、胶质母细胞瘤、脊髓星形细胞瘤和神经鞘瘤[3]。该患儿目前头部磁共振成像检查虽未见明显异常,仍需密切观察其神经系统症状进展。
XPG(OMIM 278780)是由ERCC5 基因突变引起结构特异性修复核酸内切酶XPG 活性缺陷所致,目前已报道50 余种ERCC5 基因突变与疾病相关。XPG 患者早期发病,伴急性晒伤反应,有明显的雀斑样色素沉着,可合并神经系统异常、Cockayne 综合征或脑-眼-面-骨骼综合征。其表型广泛,可从轻微到极度受累和进行性神经元死亡导致的神经系统异常[12]。虽有报道本型患者于20岁可发生皮肤癌[1],但英国一项研究提示,XPG 患者皮肤癌的发生可能少于其他XP亚型[4]。

图5 ERCC2和ERCC5蛋白结构及我国XPD和XPG型着色性干皮病患者基因突变位点 5A:ERCC2共5种突变;5B:ERCC5共8种突变。下划线指示本研究中突变
我国既往报道的5 例XPG 患者中,3 例表现为轻度表型,暂未见肿瘤发生[5,7];1 例无义突变和错义突变(p.Gln233Ter,p.Gly2Trp)患者合并恐惧症、记忆力差及感觉神经性耳聋[13];1例复合杂合无义突变(p.Gln37Ter,p.Lys392Ter)患者合并智力减退[5]。本研究中例2 表现为轻度症状,存在剪切突变c.2533+2T>C 和错义突变p.Ala818Val。尽管突变均位于核酸内切酶活性区域(I区)内(图5B),但与无义或移码突变相比,剪切突变和错义突变对蛋白的影响可能相对较小,保留了一些残留功能而导致轻症。既往研究者推测ERCC5基因的蛋白截短突变可导致XPG/Cockayne 综合征,携带至少1 个ERCC5 非截短突变的患者可能具有轻度XPG 表型[14],本研究结果支持该推测,并显示I区的错义和剪切突变亦可导致轻症表型。但有文献报道两姐弟患者核酸内切酶I 区纯合错义突变p.Leu778Pro导致轻度XP 皮肤表型合并轻微神经异常[15]。本研究中患儿年龄较小,仍需进一步观察是否出现迟发性神经系统症状。
本文提醒临床医生需提高对XP早期临床表现的认识,以期早诊断、早防治。XPG 患者早期临床表现可较为轻微;合并神经系统异常的患者除XPA外,应注意XPD 的可能。由于本病致病基因较多,临床表型复杂,单独应用Sanger 测序费时、费力且费用较高,采用高通量WES 结合Sanger 测序验证的方法可快速、准确、经济地进行基因诊断和分型,进一步指导预后及遗传咨询。
利益冲突 所有作者均声明不存在利益冲突
猜你喜欢
广西医科大学学报(2022年5期)2022-06-07
中国循证儿科杂志(2022年2期)2022-05-26
昆明医科大学学报(2022年3期)2022-04-19
种子(2021年3期)2021-04-12
中国生殖健康(2020年2期)2021-01-18
中南医学科学杂志(2019年6期)2019-12-05
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
