
国内首报Legius综合征一家系及基因突变分析
2020-06-05 06:53:52陈志明王小坡孙建方杨勇
中华皮肤科杂志 2020年4期
陈志明 王小坡 孙建方 杨勇
1中国医学科学院 北京协和医学院 皮肤病医院遗传病中心,南京210042;
2中国医学科学院 北京协和医学院 皮肤病医院病理科,南京210042
通信作者:王小坡,Email:13770757675@163.com;杨勇,Email:yyang@pumcderm.cams.cn
Legius 综 合 征(Legius syndrome,LS;OMIM 611431)临床表现与Ⅰ型神经纤维瘤病(neurofibromatosis type 1,NF1;OMIM 162200)相似,又称为类Ⅰ型神经纤维瘤样综合征(neurofibromatosis type 1⁃like syndrome),是由生殖细胞系SPRED1 基因杂合失功能突变引起的罕见常染色体显性遗传病,表现为多发咖啡斑,可伴有腋窝和/或腹股沟雀斑、巨头畸形,但缺少神经纤维瘤、视神经通路神经胶质瘤、虹膜Lisch 结节、骨骼异常等NF1 典型症状。目前国外有100 余例LS 报道[1],而国内尚未见报道,我们对仅表现为多发咖啡斑的一个家系进行基因检测,结合临床表现确诊为LS。
对象与方法
一、临床资料
先证者男,12 岁。因躯干、四肢泛发淡褐色斑12年于我院门诊就诊。患者出生时躯干即出现数个咖啡斑,逐渐扩大、增多,无自觉症状。平素体健,智力发育正常,既往史及个人史无特殊。父母非近亲结婚,先证者父母及外祖父母中仅其母亲有类似症状(图1)。体检:一般情况好,发育正常,各系统均未见异常。皮肤科检查(图2A、2B):躯干、四肢散在10余处边界清楚、不规则或椭圆形、米粒至鸡蛋大小咖啡色斑,腋窝、腹股沟可见散在浅褐色斑点,未见丘疹结节、虹膜Lisch结节。实验室检查:血、尿常规和心脏彩色多普勒超声检查及脊柱正侧位X 线和头颅MRI 检查均未见异常。先症者母亲躯干、四肢多发大小不一、不规则咖啡斑(图2C),腋窝、腹股沟散发浅褐色斑点,余无特殊所见。
二、方法
1.全外显子组测序:本研究经中国医学科学院皮肤病医院医学伦理委员会批准[(2019)临审第(005)号],患者监护人签署知情同意书。用乙二胺四乙酸抗凝管采集先证者、先证者父母及外祖父母外周血各2 ml,用Blood DNA Mini Kit(德国Qiagen 公司)提取全基因组DNA,将先证者DNA 送至北京诺禾致源生物信息科技有限公司,使用PE150 测序系统(美国Illumina 公司)进行人类全外显子组高通量测序。外显子测序后获得原始数据,通过BWA 软件进行序列比对,采用GATK和VarScan软件对变异识别,包括对SNP、InDel 等进行检测、注释和统计分析。用Annovar 软件从外部数据库得到检出的变异频率、致病信息,对检出的变异进行注释。根据分析结果,参考dbSNP、人类基因突变数据库(HGMD)、Exac等数据库找出致病突变。
2.Sanger测序验证突变:根据全外显子组测序检测到的突变,对家系各成员DNA 标本进行突变位点Sanger 测序验证。PCR 扩增家系各成员SPRED1 基因第7 外显子序列。使用PubMed Primer Blast在线设计引物,由北京擎科生物公司合成,正向引物5′⁃GTGGTATTTAAGACGCAGCC⁃3′,反向引物5′⁃CCAGAAATGGTTTCCTTCTG⁃3′。扩增条件:98 ℃预变性3 min,98 ℃变性10 s、55 ℃退火10 s、72 ℃延伸10 s共35个循环,之后72 ℃延伸2 min。PCR反应产物经琼脂糖凝胶电泳切胶纯化后由北京擎科生物公司测序。测序结果应用Chromas 软件分析,并运用DNAMAN 软件与SPRED1基因参考序列进行比对。
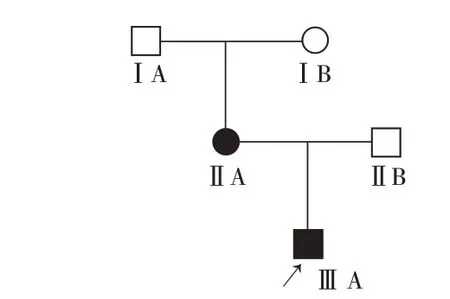
图1 Legius综合征患者家系图

图2 Legius综合征先证者及其母亲临床表现 2A:先证者躯干多发不规则或椭圆形咖啡斑;2B:先证者腋窝见咖啡斑及不典型雀斑(黄色箭头指示);2C:先证者母亲躯干多发不规则咖啡斑
结 果
先证者基因组DNA全外显子组测序结果分析显示,与多发性咖啡斑相关的NF、Noonan 综合征、Costello 综合征、LEOPARD 综合征、Cardio⁃facio⁃cutaneous 综合征和McCune⁃Albright 综合征的致病基 因NF1、NF2、PTPN11、KRAS、HRAS、NRAS、BRAF、RAF1、SOS1、LZTR1、MAP2K1、MAP2K2、MAS 均未发现致病突变,SPRED1 基因第7 号外显子自第1 220 位核苷酸起发生19 个碱基杂合缺失(NM_152594,exon7,c.1220_1238del),引起氨基酸序列发生移码突变(p.L407fs),成为候选致病突变。进一步对先证者和其父母及外祖父母进行SPRED1基因第7号外显子PCR扩增及正反向测序发现,先证者及患病母亲均携带该杂合突变,先证者外祖父母、父亲均未检出该突变(图3),该家系中该突变与疾病符合共分离。综合临床表现、病史和基因检测结果,认为SPRED1: c.1220_1238del:p.L407fs为该家系致病突变,先证者符合LS诊断。
讨 论

图3 Sanger 测序图 先证者及母亲SPRED1 基因第7 号外显子从第1 220位核苷酸起发生碱基杂合缺失(红色箭头),先证者外祖父母未检出该突变
LS 由Brems 等于2007 年首次报道,并且确定该病由SPRED1 基因失功能杂合突变引起[2]。SPRED1 蛋白由444 个氨基酸组成,包括3 个结构域,是肾素血管紧张素系统-丝裂原活化蛋白激酶(MAPK)重要抑制剂,通过调节RAS信号通路控制MAPK 信号转导参与调节细胞生长、分化和衰老,对个体正常生长发育至关重要[3]。除SPRED1基因外,其他相关调控基因突变导致肾素血管紧张素系统-MAPK信号转导异常后可引起一组遗传性综合征群,因临床表型均可出现颅面畸形、心血管系统异常、皮肤和神经系统症状,又称之为神经-心脏-面部-皮肤综合征,包括NF1、LS、Noonan 综合征、Costello 综合征、cardio⁃facio⁃cutaneous 综合征和LEOPARD综合征[4]。
LS 临床表现为多发咖啡斑,部分可有腋窝和腹股沟区雀斑、眼距增宽、巨头畸形、脂肪瘤,患者可伴有轻度学习障碍,但不发生神经纤维瘤、视神经胶质瘤和虹膜Lisch 结节[1,5]。LS 需与NF1 鉴别诊断,据估计,高达5%符合美国国立卫生研究院诊断NF1 标准的患者实际为SPRED1 基因突变导致的LS,而非NF1[6]。对现有报道病例分析表明,LS并发症的严重程度以及并发恶性肿瘤概率均低于NF1[1],因此早期对这两种疾病进行鉴别诊断对预后判断、遗传咨询和产前诊断有重要意义。LS 与NF1早期均表现为多发咖啡斑,而NF1特异性表现需要到一定年龄才发生,所以通过临床症状早期鉴别诊断较为困难,当皮肤科医生接诊仅有多发性咖啡斑伴或不伴腋窝雀斑但缺少NF1 其他相关症状的患者时,应考虑到LS 可能并可通过基因检测进行早期鉴别诊断。对患者进行检测时需注意的是,NF1 和SPRED1 基因均易发生大片段缺失[7],而目前全外显子组捕获高通量测序方法无法检出大片段缺失,因此,当高度怀疑NF1和LS而采用全外显子组测序未能发现NF1 或SPRED1 基因致病突变时,须采用多重链接探针扩增技术(multiplex ligation dependent probe amplification)排除大片段缺失可能。若基因检测明确有NF1致病突变,则需严格定期至有处理NF1经验的医院随访,重点监测患者实体瘤如视路胶质瘤、恶性周围神经鞘膜肿瘤的发生,骨骼系统异常和神经系统异常[8],而如果检出SPRED1 致病突变,家长可自行观察,重点关注患者生长发育以及学习情况。LS患者一般不需特殊治疗,如果咖啡斑影响美观可选择激光治疗。
本文先证者的母亲亦有类似症状,基因检测也检出与先证者同一突变(SPRED1: exon7:c.1220_1238del:p.L407fs),先证者外祖父母和父亲均未检出该突变,证实该突变为新发突变,该新发突变在千人基因组数据库、HGMD、ExAC EAS数据库以及诺禾致源正常人外显子突变数据库(NovoDb_WES)中均未见报道。截至目前,HGMD已收录SPRED1 基因致病突变72 个(http://www.hgmd.cf.ac.uk/ac/gene.php?gene=SPRED1),主要突变类型为小片段缺失/插入(34/72)、错义/无义突变(27/72),其余为大片段缺失和剪切突变。从目前报道的病例分析,尚未能发现基因型-临床表型明显相关性,SPRED1基因多个外显子甚至整个基因缺失患者与仅有点突变患者的临床表型相比无差别,较为特殊的1 例患者出现了腭裂,该患者染色体出现大片段缺失(6.6Mb),缺失片段包含SPRED1 以及另外29 个注释参考基因[9],既往研究者认为这些注释参考基因异常可引起唇腭裂与发育迟缓[10]。因此,还需要更多LS 患者临床资料和突变数据来分析LS基因型-临床表型相关性。
利益冲突 所有作者均声明不存在利益冲突
猜你喜欢
临床输血与检验(2022年3期)2022-06-22 02:52:50
种子(2021年3期)2021-04-12 01:42:22
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
分忧(2017年6期)2017-06-07 07:51:58
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
湖南大众传媒职业技术学院学报(2015年6期)2015-12-01 03:21:19
伴侣(2015年7期)2015-07-16 05:32:56
科技视界(2015年9期)2015-04-07 10:02:28
重庆医学(2015年12期)2015-03-05 05:52:54
