
人乳头瘤病毒58型E6E7融合基因重组巨细胞病毒的构建及鉴定
2020-05-08 04:43:32杜阳阳苗智颖李淑英郑春阳李劲涛
中国人兽共患病学报 2020年4期
杜阳阳,张 放,苗智颖,赵 红,马 迪,王 静,李淑英,郑春阳,李劲涛
宫颈癌发病率在世界范围内居女性恶性肿瘤的第二位,是危及全球妇女健康的严重疾病,据国际癌症中心(IARC)估计,全球每年宫颈癌新发病例数约52.8万,死亡人数约26.6万[1]。研究证明,人乳头状瘤病毒 (human papillomavirus,HPV) 中13种高危型长期反复感染是宫颈癌发生的必要条件[2-4];有研究表明HPV 58型在我国感染率较高[5-6]。尽管Gardasil 9 (HPV9价疫苗)已经在世界范围内上市,该疫苗可预防9型HPV所致某些疾病(包括HPV型16,18,31,33,45,52和58型所致宫颈癌、外阴癌、阴道癌和肛门癌,以及HPV 6或11型所致尖锐湿疣)[7-8],然而这种疫苗只适用于没有感染过HPV的人群,对于已经感染上述相应型别HPV而导致的病变患者没有作用,因此,研制HPV58型治疗性疫苗是非常必要的。依据上述事实和原理,本项研究以HPV58突变型E6E7(Mutant type E6E7,mE6E7)融合基因为靶点,用HPV58野生型E6E7(Wild type E6E7,wE6E7)融合基因片段作对照,利用对人无致病性的巨细胞病毒株(Towne株)细菌人工染色体(SW102 Towne bacterial artificial chromosome,SW102-T-BAC)为载体,构建携带HPV58 mE6E7及wE6E7融合基因的重组巨细胞病毒,探讨HPV58mE6E7的转化活性,为HPV58型治疗性疫苗研制奠定基础。
1 材料与方法
1.1材料 GalK质粒(pGalK), 对人无致病性的巨细胞病毒株(Towne株)细菌人工染色体(SW102 Towne bacterial artificial chromosome,SW102-T-BAC)包含巨细胞病毒的全基因组,并带有氯霉素抗性基因,为美国罗格斯大学,新泽西医学院朱桦教授惠赠。
1.2 方法
1.2.1HPV58 E6和E7野生型与突变型融合基因片段的合成 HPV E6和E7基因特有的Cys-x-x-Cys基序具有锌指结构与抑癌基因p53,Rb结合并降解抑癌基因。为消除E6,E7基因可能的恶性转化活性,将锌指结构进行突变[9-10],构建E6E7突变型融合基因。根据GenBank提供的基因编码区序列, 对野生型基因序列加以改造,同时将E7与E6之间的终止子进行改造,使其形成连续编码序列,而形成融合基因(具体改造方法见图1),这样mE6E7融合基因保证了读码框的正确性,既可消除E6与E7的转化活性又能保留其抗原性。委托北京赛思莱博科技有限公司合成突变型E6E7(mE6E7)与野生型E6E7(wE6E7)融合基因。
1.2.2带有T-ORF75左/右同源臂GalK、HPV58 wE6E7与mE6E7融合基因引物的设计及片段扩增
1)依据GenBank提供的Towne株(FJ616285.1)基因序列,分别设计其开放读码框ORF75(Towne-ORF75左/右同源臂GalK,HPV58 E6E7融合基因引物(见表1)。委托北京赛思莱博科技有限公司引物合成。2)用所设计引物,以质粒GalK、HPV58 wE6E7及mE6E7融合基因片段为模板,分别扩增GalK片断、野生型及突变型融合基因wE6E7与mE6E7片段,反应体系 (100 μL):PCR mix包括Buffer、dNTP Mixture及Taq DNA polymerase,primers 10 μmol/L (表1 GalK Primers及HPV58 E6E7 Primers), 和 200 ng GalK质粒DNA。 PCR反应循环:95 ℃/15 min; 31个循环95 ℃/30 s;55 ℃/30 s;72 ℃/1.5 min; 延伸72 ℃/10 min,4 ℃储存。用1.2%琼脂糖凝胶电泳检测PCR产物,并切胶纯化回收。
表1 本项实验中所需引物名称、序列及大小
Tab.1 the name, sequence and size of the primers required in this experiment

引物名称序列产物大小/bpGalk Primers GalK ForwardcaaccaccacctggatcacgccgctgaacccagcggcgcggccgcgctatgCCTGTTGAC-AATTAATCATCGGCA GalK Reversegctgaggagacagacggcgaggacgatgaggtaggaggggaggcctggccg-TCAGCACT-GTCCTGCTCCTT1 400HPV58E6E7 Primers HPV58E6E7 Forwardcaaccaccacctggatcacgccgctgaac-ccagcggcgcggccgcgctatgATGTTCCAGGACGCAGAGGAG HPV58E6E7 Reversegctgaggagacagacggcgaggacgatgaggtaggaggggaggcctggccg TCCTTGCTG-TGCACA GCTAG861Galk Check Primers Check Forwardgcgacttttgtaacccgtag Check Reversegcagtaggtgaaacgctttg1 550HPV58E6E7 check Primers Check Forwardgcgacttttgtaacccgtag Check Reversegcagtaggtgaaacgctttg1 012
1.2.3将已纯化的Galk片段插入Towne开放读码框75(T-ORF75) 将上述纯化的GalK片段电转到SW102-T-BAC感受态细胞,然后用1 mL LB培养液,于32 ℃振摇1 h后,离心,再用1×M9液洗涤沉淀物2次,将洗涤后的菌均匀涂布于M63配制的含有Galk的培养基[7],在32 ℃培养3 d后长出细菌克隆;挑选3个克隆,提取质粒DNA,PCR检测GalK(表1 GalK Check Primers)。检测正确克隆质粒命名为SW102-T-ORF75-GalK-BAC。并制备其电转感受态细胞,负80 ℃储存备用。
1.2.4HPV58E6E7融合基因代替GalK基因 取约800 ng已纯化的T-ORF75左/右同源臂HPV58 wE6E7与mE6E7融合基因片段,分别电转化到SW102-T-ORF75-GalK-BAC感受态细胞,用1 mL LB培养液,于32 ℃振摇4 h 30 min,离心,再用1×M9液洗涤细菌沉淀物2次,将细菌沉淀物混悬于1 mL M9洗涤液后,分别在进行10倍及100倍稀释后,用不同浓度的稀释菌液涂布于脱GalK培养基[11],32 ℃培养3 d,有克隆形成;分别选2个克隆,提取质粒DNA,PCR检测(表1 HPV58E6E7 Check Primers),正确克隆命名为SW102-T-ORF75-HPV58-mE6E7-BAC,SW102-T-ORF75-HPV58-wE6E7-BAC。
1.2.5转染 ARPE-19细胞接种于12孔细胞培养板,常规培养,待ARPE-19细胞生长到汇合度为60%时,用脂质体介导分别将SW102-T-BAC、SW102-T-ORF75-HPV58-mE6E7-BAC与SW102-T-ORF75-HPV58-wE6E7-BAC质粒转染ARPE-19细胞,每种质粒转染3个复孔,剩余培养板中的3个孔细胞作为对照。24 h后将细胞培养液更换;并且每天观察细胞生长状况。待细胞长满各孔后,转移到培养皿(直径为10 cm)继续培养。
1.2.6转染细胞中HPV58 mE6E7与wE6E7表达状况 待10 cm培养皿中细胞完全汇合后,收集各组细胞,分别提取各组细胞RNA,逆转录PCR扩增检测所收集细胞中HPV58 mE6E7与wE6E7的表达,并将PCR产物测序分析。
1.2.7软琼脂克隆实验 底层软琼脂制备:配制1.5%琼脂10 mL高压灭菌,待其冷却至大约50 ℃,加入2XDMEM 10 mL,混均,于24孔板的每个孔中加入0.8 mL,于室温使其凝固。
上层琼脂制备:将10 mL已灭菌含0.7%琼脂冷却至大约50 ℃,迅速加入10 mL含20%血清的2XDMEM培养液和稳定表达SW102-T-BAC、SW102-T-ORF75-HPV58-mE6E7-BAC与SW102-T-ORF75-HPV58-wE6E7-BAC及未转染的ARPE-19对数生长期细胞(浓度为3×103个/mL),混匀,浇入底层铺有软琼脂的24孔培养板中,每组细胞接种6个复孔,琼脂凝固后将培养板放置在5% CO2、温度为37 ℃及饱和湿度环境下培养12 d。每天在倒置显微镜下观察软琼脂中细胞克隆形成情况,并记录克隆形成数量,以分裂3次增殖8个细胞作为1个克隆,计算克隆形成率。克隆形成率(%)=(克隆数/接种细胞数)×100%。
2 结 果
2.1HPV58 E6和E7野生型与突变型融合基因片段的合成 将HPV58 E6蛋白降解P53的活性部位、E7蛋白与pRB结合位点关键氨基酸改造后,所得片段基因序列如图1。
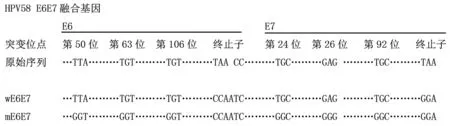
图1 HPV58 E6E7野生型与突变型融合基因突变位点序列Fig.1 HPV58 E6E7 fusion gene sequence of Wild-type and Mutant type
2.2GalK插入SW102-T-ORF75-BAC GalK培养基中培养3 d后,选取其中3个菌落,提取质粒DNA,以3个菌落质粒DNA为模板PCR扩增,检测到大小为 1 550 bp的PCR产物(图2),所得产物大小与预期结果相一致,表明GalK已克隆至Towne细菌人工染色体的开放读码框75,将正确克隆命名为SW102-T-ORF75-GalK-BAC。

M为1kb plus DNA ladder,1为阴性对照,2-4为GalK插入SW102-T-ORF75-BAC后,所选取的GalK培养基中生长的3个克隆,以所选的3个克隆质粒DNA为模板,PCR产物电泳的结果。图2 GalK克隆到SW102-T-ORF75-BAC后PCR扩增检测结果Fig.2 Results of PCR amplification for detection GalK cloned to SW102-T-ORF75-BAC
2.3HPV58E6E7融合基因代替galK基因 经过脱GalK培养基培养后,分别选取2个菌落,检测到大小为1 012 bp的PCR扩增产物(图3),所检测到的片段与预期结果一致,证实HPV58 wE6E7与mE6E7分别克隆至SW102-T-ORF75-GalK-BAC。正确克隆命名为SW102-T-ORF75-HPV58-wE6E7-BAC,SW102-T-ORF75-HPV58-mE6E7-BAC。

M为1kb plus DNA ladder,1是阴性对照,2-3 :HPV58-mE6E7替代GalK所选2个克隆PCR扩增后电泳结果,4-5:HPV58 wE6E7替代GalK所选2个克隆PCR扩增后电泳结果。图3 HPV58 mE6E7与wE6E7插入SW102-T-ORF75-GalK-BAC替代GalK所选克隆PCR扩增后电泳结果Fig.3 Results of PCR amplification for verification galk replaced by HPV58-mE6E7与wE6E7 in SW102-T-ORF75-GalK-BAC
2.4转染与未转染细胞生长状况 分别将 SW102-T-BAC、SW102-T-ORF75-HPV58-mE6E7-BAC与SW102-T-ORF75-HPV58-wE6E7-BAC质粒转染ARPE-19细胞,培养10 d 后,在倒置显微镜下观察,转染SW102-T-ORF75-wE6E7-BAC的细胞生长失去接触抑制,出现重叠生长现象,其形态由原来长梭形变为变圆、肿胀、胞浆颗粒增多(图4)。

A为未转染细胞; B为转染SW102-T-BAC至ARPE-19的细胞; C为转染SW102-T-ORF75-HPV58-mE6E7-BAC至ARPE-19的细胞;D为转染SW102-T-ORF75-HPV58-wE6E7-BAC至ARPE-19的细胞。图4 转染ARPE-19细胞生长状况(倒置显微镜下观察结果 ×200)Fig.4 Growth of ARPE-19 cells were transfected (The results were observed under an inverted microscope ×200)
2.5转染与未转染细胞中T-ORF75-HPV58-mE6E7与T-ORF75-HPV58-wE6E7表达状况 第20 d 收集转染后细胞,分别提取各组细胞RNA,逆转录PCR检测T-ORF75-HPV58-mE6E7与T-ORF75-HPV58-wE6E7 表达状况,逆转录PCR检测到转约250 bp、1 012 bp及1 012 bp PCR产物,分别为转染SW102-T-BAC、SW102-T-ORF75-HPV58-mE6E7-BAC及SW102-T-ORF75-HPV58-wE6E7-BAC的结果(图 5)。 测序分析表明HPV58-mE6E7与HPV58-wE6E7稳定表达于转染的细胞中。
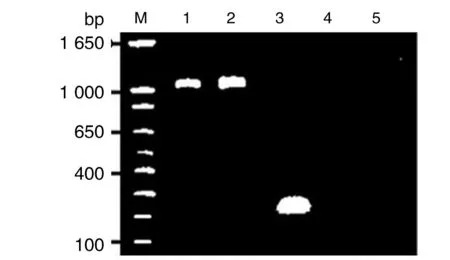
M为1kb plus DNA ladder,1:将SW102-T-ORF75-HPV58-mE6E7-BAC转染 ARPE-19细胞,培养后提取细胞RNA,逆转录PCR扩增产物电泳的结果;2:将SW102-T-ORF75-HPV58-wE6E7-BAC 转染 ARPE-19细胞,培养后提取细胞RNA,逆转录PCR产物进行琼脂糖凝胶电泳的结果;3:用SW102-T-BAC转染细胞ARPE-19,将转染的细胞培养后提取RNA,然后逆转录PCR扩增产物电泳的结果,4:将未转染细胞提取的RNA,进行逆转录PCR扩增,其产物电泳的结果;5:是阴性对照。图5 细胞中T-ORF75-HPV58-mE6E7与T-ORF75-HPV58-wE6E7逆转录PCR检测结果Fig.5 Results of reverse transcription PCR of T-ORF75-HPV58-mE6E7 and T-ORF75-HPV58-wE6E7 in transfected cells
2.6软琼脂克隆实验 以分裂3次增殖8个细胞作为一个克隆,培养至第6 d,在倒置显微镜下可观察到稳定表达T-ORF75-HPV58-wE6E7-BAC的细胞组有克隆形成,而稳定表达T-ORF75-HPV58-mE6E7-BAC、T-BAC及未转染的ARPE-19细胞未见克隆形成;培养至12 d,克隆明显增多增大,细胞呈堆积生长(图6),克隆形成率为23.5%(47/200)。稳定表达T-ORF75-HPV58-mE6E7-BAC、 T-BAC、及未转染的ARPE-19细胞组未见有克隆形成。
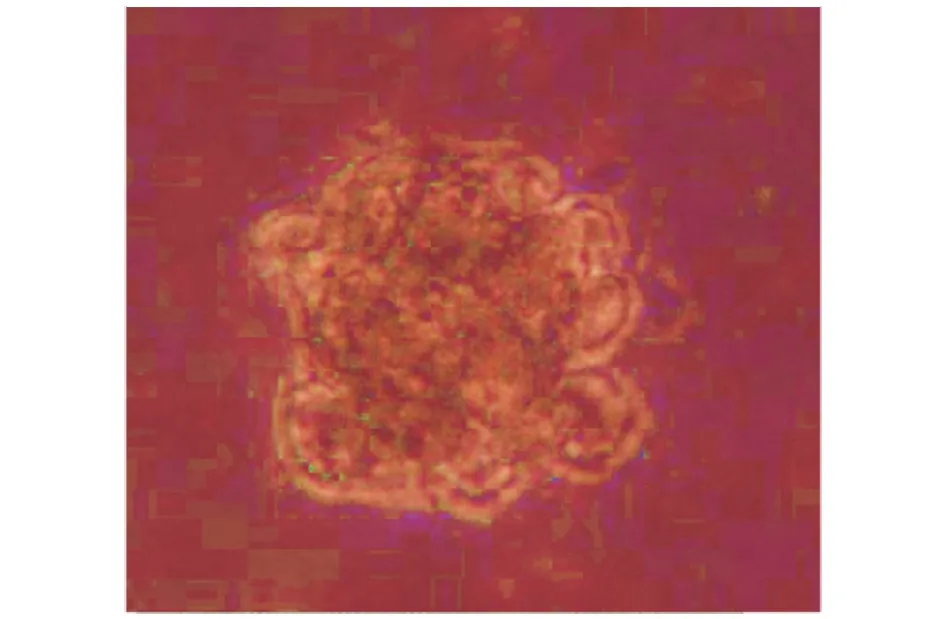
图6 转染SW102-T-ORF75-HPV58-wE6E7的ARPE-19细胞在软琼脂中形成的典型克隆(×200)Fig.6 Clone of ARPE-19 cells were transfected with SW102-T-ORF75-HPV58-wE6E7 in soft AGAR(×200)
3 讨 论
虽然世界范围内已有HPV预防性疫苗上市[8,12],预防HPV疫苗可保护健康人群免患相应型别HPV感染所导致的疾病,但对已感染相应HPV型别患病的人群没有治疗的效果,对已感染各型别HPV导致的各类疾病,还需相应的治疗性疫苗。目前治疗性HPV疫苗还处于研发阶段,理想治疗性HPV疫苗既要在患者体内产生强的细胞免疫,又要清除患者体内已感染的HPV,同时还要在患者体内产生中和抗体,中和患者体内病毒,而抑制病毒的传播。
高危型HPV感染的恶性肿瘤中,E7与E6蛋白是形成HPV相关恶性肿瘤及维持其恶性表型所必需的蛋白;E6通过降解P53,降低细胞对DNA合成修复能力,容易出现错误的基因结构;E7与pRb基因结合,诱导中心体合成异常增多,使得有丝分裂异常紊乱,非整倍体细胞出现,从而导致细胞的恶性转化[13-14]。E6和E7两个癌基因在HPV阳性恶性肿瘤细胞中持续表达,而在正常细胞中不表达,因此,E6和E7蛋白属于肿瘤细胞中特异性的病毒抗原,是HPV阳性肿瘤免疫治疗的理想靶抗原,可将HPV E6和E7基因加以改造研制成治疗性疫苗;但如果用野生型E7与E6基因直接作抗原,构建E6E7融合基因,有潜在致癌性;因此,需将野生型E7基因与pRB基因结合位点加以改造,E6降解P53基因位点加以改造,使其发生突变,敲除E7与E6基因的转化活性,并将E7与E6各自的终止密码子加以改造,这样所构建的突变型融合基因,既保留了其抗原性,又去除了其致癌性,同时,也保证了融合基因读码框的正确性。因此,本项研究将E6基因结合p53、E7结合Rb活性部位锌指结构域内一些氨基酸序列加以改变,消除E6结合p53、E7结合Rb基因的降解活性,即E6第50位的亮氨酸在高危HPV中高度保守,可能在E6转化过程中起重要作用;第63位和第106位的半胱氨酸位于E6的一对锌指结构的两侧,在对p53的结合中起重要作用。E7中第24位半胱氨酸和第26位谷氨酸位于一对锌指结构的两侧,第92位半胱氨酸位于一个独立的锌指结构中[10],因此,我们将这些部位碱基序列加以改变,消除E6终止密码子,并保证E6与E7读码框的正确性,使其形成融合基因,分别构建了HPV58mE6E7和HPV58wE6E7融合基因的重组病毒。通过转染细胞形态学观察及软琼脂克隆实验证实所构建的重组病毒T-ORF75-HPV58-mE6E7已消除致癌活性。
利用Towne细菌人工染色体为载体,通过同源重组,已成功将HPV58E6E7融合基因连接到Towne的ORF75;Towne的ORF75编码包膜糖蛋白gH,具有免疫原性,在诱导宿主免疫反应方面发挥着重要作用。今后将进一步探讨重组病毒T-ORF75-HPV58-mE6E7的免疫原性、免疫反应性及其作为治疗性疫苗的免疫效果。
本项研究采用巨细胞病毒减毒活疫苗Towne株[15-16]细菌人工人色体为载体,构建表达HPV58E6E7去除其转化活性融合蛋白的重组巨细胞病毒,并转染ARPE-19细胞,结果显示已成功构建了携带Towne-ORF75-HPV58 mE6E7与wE6E7基因的重组病毒,为HPV58型治疗性疫苗的研究奠定了基础。
利益冲突:无
猜你喜欢
新农业(2021年9期)2021-06-20 11:26:32
波谱学杂志(2021年1期)2021-03-12 07:37:28
科学(2020年3期)2020-11-26 08:18:34
武昌理工学院学报(2017年1期)2017-05-11 14:19:39
食品与生物技术学报(2017年2期)2017-04-09 11:43:29
生物学教学(2016年9期)2016-08-21 02:37:00
考试周刊(2015年91期)2015-09-10 07:22:44
安徽医药(2014年9期)2014-03-20 13:14:09
吉林大学学报(医学版)(2014年2期)2014-02-27 06:48:05
安徽医科大学学报(2014年11期)2014-02-15 10:20:36
