
3p25.3p25.2 染色体杂合缺失伴矮小及多发畸形1 例遗传学特征与临床表型分析
2020-04-04 10:50:28赵晓峰毛国顺刘德云王宁玲邱文娟
临床儿科杂志 2020年3期
赵晓峰 毛国顺 李 利 刘德云 王宁玲 邱文娟
1.安徽医科大学第二附属医院儿科(安徽合肥 230601);2.阜阳市人民医院儿科(安徽阜阳 236000);3.上海交通大学医学院附属新华医院内分泌遗传代谢研究室(上海 200092)
染色体异常是导致儿童生长发育迟缓、智力低下、运动行为发育障碍及多发畸形的常见原因之一。3号染色体p25.3p25.2(3p25.3p25.2)染色体缺失综合征(简称3P综合征)是一组染色体缺失疾病[1-2],主要表现为全面生长发育迟缓、智力低下、小头畸形、小下颌、长人中、上睑下垂、多指畸形、肌张力减低等,临床亦可见先天性心脏病、刻板动作、胃肠道畸形,肠旋转不良等。目前国外报道50 余例,国内病例更少。合并先天性十二指肠闭锁者,国内外尚未见报道。现总结分析于2018—2019 年多次就诊于安徽医科大学第二附属医院及阜阳市人民医院确诊3 p 25.3 p 25.2 杂合缺失综合征男童的临床资料,分析其临床表型及分子遗传学特征。
1 临床资料
患儿,男,1岁4个月,生后曾因“先天性十二指肠闭锁、肠旋转不良”在外院行腹腔镜探查Ladd手术加肠切除吻合术。患儿系G1P1,孕36+5周自然分娩。出生体质量2 450 g,出生身长48 cm。目前运动发育行为落后,不会翻身,不能独坐,反应迟钝,不会逗笑,不会发出有意义的言语,四肢肌张力低,生长发育明显落后(图1)。婴儿期有喂养困难及睡眠障碍。父母均体健,否认遗传性疾病家族史。既往曾在外院确诊先天性心脏病及甲状腺功能减低症,一直口服左旋甲状腺素治疗。外院阴囊超声检查:双侧隐睾伴双侧精索鞘膜积液。体格检查:身长64 cm(<P3),体质量5.4 kg(<P3),头围42.5 cm(<P3);神志清,呼吸快,50次/min,双肺底可闻及细湿啰音;小头畸形,双眼距增宽,双眼睑无下垂,无内眦赘皮,宽鼻桥,长人中,小下颌,耳位低,双侧耳前瘘管(图2);心率120次/min,胸骨左缘Ⅱ~Ⅲ肋间可闻及3/6级收缩期杂音,上腹部可见一长约10 cm 纵行手术瘢痕;四肢肌张力低,双手无通贯手,无多指(趾)畸形,有隐睾,龟头裸露;双侧腹股沟区有斜疝,无嵌顿。实验室检查:肝肾功能、电解质均无异常,血串联质谱无异常。染色体46,XY。6月龄复查超声心动图示动脉导管未闭、卵圆孔未闭。
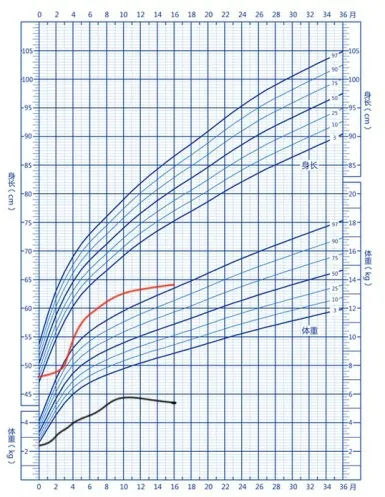
图1 患儿身长、体质量生长资料曲线图
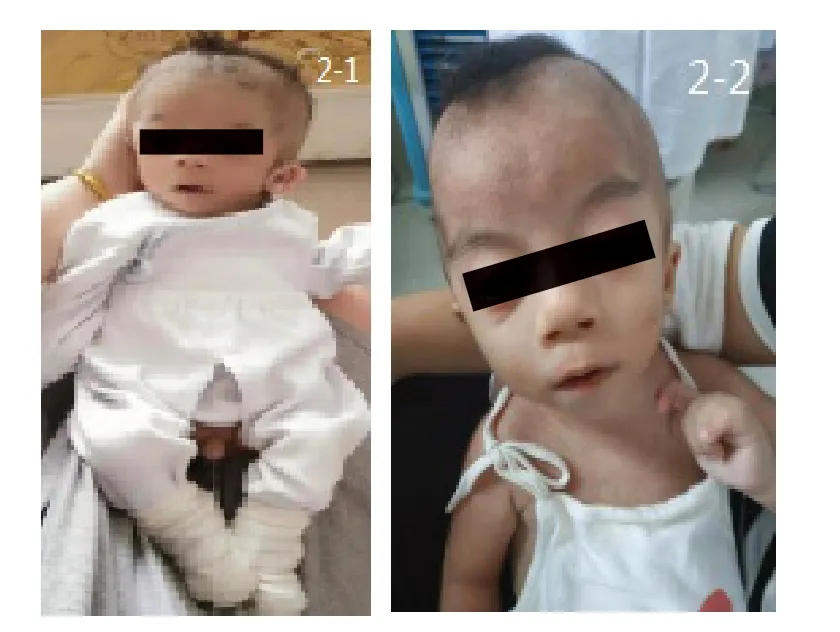
图2 患儿特殊面容
因患儿明显营养不良、全面生长发育落后、特殊面容伴多发畸形,经医学伦理审核及家长知情同意行全外显子测序检查。应用安捷伦外显子芯片捕获加高通量测序,通过对该样本特殊计算,发现存在染色体基因拷贝数变异。在3号染色体:9470607-12545298区域可能存在约3 074 kb杂合缺失突变(图3)。
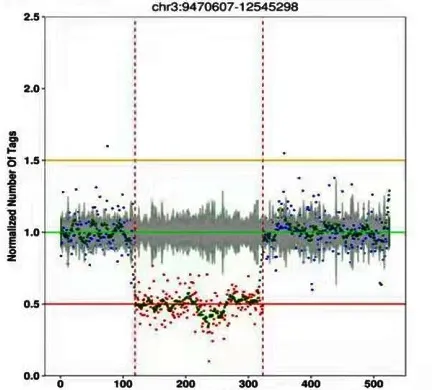
图3 全外显子测序特殊计算结果
因二代测序并非CNV 检测金标准,为明确该段杂合缺失是否真实存在,再次行基因芯片检查。采用Affymetrix 公司标记检测试剂盒及相关产品,使用cytoscan 750 k 芯片,经过DNA 的提取和质控、酶切及标记、杂交纯化及扫描和分析等相关步骤,检测结果:arr[GRCh37]3p25.3 p25.2 (9223245-12550647)×1,在受检者的3 号染色体p 25.3 p 25.2 区域存在一段大小约3 327 kb杂合性缺失(图4)。该段基因缺失覆盖多个表型明确的OMIM 基因如SETD 5、VHL、FANCD 2等,根据美国医学遗传学与基因组学学会(ACMG)标准为致病性变异。SETD 5 单倍剂量不足可明确导致常染色体显性遗传性智力落后;VHL单倍剂量不足明确导致Von Hippel-Lindau 综合征(OMIM193300),为类癌易感综合征。结合本例患儿有低出生体质量、宫内发育迟缓、明显矮小、智力低下伴特殊面容、肌张力减低、隐睾、喂养困难、睡眠障碍,伴先天性心脏病、甲状腺功能减退症、先天性十二指肠闭锁及肠旋转不良,SNP 微阵列芯片证实3p25.3-p25.2区域存在一段大小约3 327 kb杂合性缺失,证实为3p25.3p25.2缺失综合征。
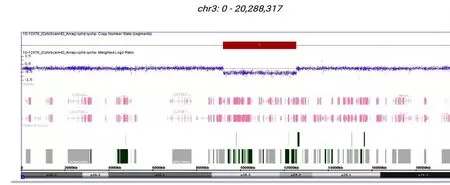
图4 基因芯片验证结果
2 讨论
3p25.3p25.2缺失综合征可出现矮小、智力低下、特殊面容、多发畸形及多脏器发育异常各种各样的临床表型[3]。该病最早于1978年首次报道[4]。根据染色体片段缺失的位置和大小可有不同的临床表型[5]。目前全世界报道共50余例,国内病例更少。据文献报道,3 p 25.3 p 25.2 缺失综合征可有胃肠道畸形,先天性十二指肠闭锁国内外均未见报道。本例患儿合并先天性十二指肠闭锁为首次发现,丰富了3p25.3p25.2缺失综合征的临床表型。
DECIPHER 数据库显示本例患儿该段染色体片段缺失共包含39个基因缺失。SETD5、SRGAP3、VHL、FANCD 2、SEC 13、GHRL、PPARG、CIDEC、HRH 1、SYN2、SLC6A1、ATg7、BRPF1、CRELD1、OGG1等15个基因与本例患儿临床表型关系密切。其中SETD5、VHL及FANCD2基因均是OMIM数据库收录的有明确报道的致病性变异[6]。
3 p 25.3 p 25.2 缺失综合征患者均具有身材矮小、全面生长发育落后等临床表型。文献报道CHL、CALL、CNTN4、SRGAP3等多个基因在3p25.3p25.2缺失综合征患儿生长发育迟缓中起重要作用[7]。导致本例矮小、生长发育落后的因素有:①先天性十二指肠闭锁及肠旋转不良。3 p 25.3 p 25.2 缺失综合征患儿肠旋转不良或食管闭锁等胃肠道发育异常[8],可能与SEC13基因缺失导致其表达的Secl3蛋白的缺失相关。SEC13基因在肠道上皮发生、视网膜、颅面部发育及消化器官发育方面均起重要作用。②婴儿期喂养困难。其机制为GHRL基因缺失影响Grelin释放[9],摄食行为、能量代谢失调而导致严重的喂养困难,生长发育落后。③PPARG基因和细胞死亡诱导的DFF 45 样效应子3 基因(CIDEC)对脂肪细胞分化和能量储存方式的影响。PPARG基因位于3p25,调节脂肪细胞分化,参与脂肪细胞凋亡,PPARG基因缺失可致脂肪萎缩[10]。CIDEC基因缺失使脂肪间充质干细胞丧失脂肪分化能力,从能量储存转变为能量消耗,加重营养不良及生长发育迟缓[11]。④合并甲状腺功能减退症。曾有报道3 p 25.3 pter 末端缺失患者存在甲状腺功能减退症[4,12]。⑤睡眠障碍。文献报道与HRH 1基因缺失有关[13]。HRH1基因缺失,其表达的Hrh1蛋白亦缺失,可有严重睡眠障碍。⑥3p25.3p25.2缺失综合征患儿可合并心脏及骨骼异常。
智力发育落后是3 p 25.3 p 25.2 缺失综合征较重要的临床表型,SETD5及SRGAP3基因可导致患儿智力发育低下。SETD5基因位于3p25.3,通过形成大的多蛋白复合物来修饰或重组染色体,在基因转录的过程中起组蛋白乙酰化调节作用,已被证实可致常染色体显性遗传智力低下[14-16]。SRGAP3基因在颅脑发育时神经轴突中高表达,SRGAP3缺失可导致严重的智力发育迟缓[17]。
癫痫发作是3 p 25.3 p 25.2 缺失综合征的常见临床表型。研究证实与SYN2、SLC6A1及ATg7等基因缺失相关。SYN2基因参与神经递质的释放和突触小泡的形成,与额叶突触形成有关,有神经特异性,SYN 2基因变异可导致癫痫发生[18]。SLC6A1基因在人和哺乳动物发育期和成熟大脑高表达,其编码的GABA转运体GAT-1可从突触间隙重摄取GABA,该基因缺失可致癫痫发作[19]。ATg 7基因参与自噬过程,敲除ATg 7基因的小鼠显示出自噬行为完全丧失,包括异常抓握反射、震颤、运动失调、协调能力丧失、刻板行为及帕金森样表现[20]。
特殊面容及多发畸形在3 p 25.3 p 25.2 缺失综合征中较常见,可能与BRPF1基因和Secl3基因密切相关。BRPF1基因在维持正常的颅面骨发育必需的HOX基因表达方面有重要作用[21]。Secl 3基因缺失则可导致骨胶原转运功能受损或缺陷,是引起颅面部发育缺陷、视网膜病变、特殊面容和肠道发育异常[22]。
CRELD1基因位于3p25,是第1个被发现的与先天性房室间隔缺损发病相关的基因[23]。CRELD1基因可反向调节转录因子Sox9和聚集蛋白聚糖Aggrecan的表达水平,与心内膜垫缺损发生和房室间隔缺损有关[24-26]。
本例患儿缺失染色体片段包括VHL、OGG 1、FANCD 2等常见的抑癌基因。VHL位于3 p 25.3,与Von Hippel-Lindau 综合征有关[27],VHL基因缺失可导致肿瘤高发[28-29];OGG1基因位于3p25p26,表达的OGG 1 蛋白失活或人工敲除时,可降低DNA 修复功能,导致高肿瘤易感性[30];OGG1蛋白能维持抑癌基因P53的稳定性,协同抑制肿瘤发生,OGG1基因缺失与皮肤癌、乳腺癌、前列腺癌的发生率相关。FANCD2基因是范可尼贫血通路的关键基因,与乳腺癌、口腔癌、卵巢癌、肺癌和黑色素瘤的发病相关[31]。该患儿多种抑癌基因缺失,恶性肿瘤倾向高,不建议生长激素治疗。同类文献均未见应用生长激素的相关报道,故仅予营养支持,纠正甲状腺功能减退症。本例尚未观察到癫痫、肾脏异常及听力丧失等,需后续观察相应表现。
综上,3p25.3p25.2缺失综合征根据染色体片段缺失的位置及大小,临床表现差异较大。本例患儿丰富了3p25.3p25.2缺失综合征的临床表型,对有特殊面容、多发畸形等临床表现者,应考虑染色体疾病,及时行染色体、基因芯片等检查明确,以提高患儿的生活质量。本例患儿后续将密切关注某些尚未出现的临床表现如癫痫、癌症高发倾向等。
猜你喜欢
心电与循环(2021年4期)2021-11-29 02:41:56
天津医科大学学报(2021年4期)2021-08-21 02:14:52
心肺血管病杂志(2020年5期)2021-01-14 00:43:30
心电与循环(2020年3期)2020-06-18 13:43:12
现代园艺(2017年21期)2018-01-03 06:41:32
中国眼镜科技杂志(2016年17期)2016-10-24 08:36:30
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
中国当代医药(2015年30期)2015-03-01 02:08:19
现代检验医学杂志(2015年5期)2015-02-06 01:42:20
