
戊二酸血症I 型患儿家系基因变异检测及产前诊断
2020-04-04 10:50:28马定远
临床儿科杂志 2020年3期
陆 璐 马定远 成 建
1.江苏卫生健康职业学院生化教研室(江苏南京 211800);2.南京医科大学附属妇产医院产前诊断中心(江苏南京 210004)
戊二酸血症I型(glutaric aciduria type I,GA I)是一种少见的神经退行性疾病,为常染色体隐性遗传,全球发病率约为1/110000[1]。患儿最早可于1 月龄时出现症状,临床表现多样,主要为头大畸形、肌张力异常、运动障碍等[2-4],常在炎症、发热、呕吐、腹泻、疫苗接种、手术等应激状态下诱发。目前研究认为,GA I 是由于戊二酰辅酶A 脱氢酶(glutaryl CoA dehydrogenase,GCDH)活性降低或缺失导致戊二酸、3-羟-戊二酸、戊烯二酸及戊二酰肉碱等有机酸在组织及体液内异常蓄积造成神经系统损伤所致[5]。新生儿血液、尿液质谱筛查可早期诊断GA I。患儿须在出现症状前限制赖氨酸、色氨酸的摄入,并且长期坚持饮食控制方案[6],这必然会给患儿及家庭造成严重的心理、生理及经济负担。因此对于有再生育要求的患儿家庭,胎儿产前分子遗传学诊断尤为重要。本研究对1 个GA I 家系进行基因检测,以明确致病变异,并为患儿家庭再生育提供遗传咨询和产前诊断。
1 临床资料
患儿(先证者),男,4岁。6月龄不能抬头、不能抓物。8月龄时当地医院行尿液气相色谱-质谱(GC/MS)检测提示戊二酸浓度明显升高(118.6 μmol/L,参考值0~4.0 μmol/L);血高效液相色谱-串联质谱(LC/MS)检测提示戊二酰肉碱浓度明显升高(0.51 μmol/L,参考值0~0.20 μmol/L),游离肉碱浓度低(5.67 μmol/ L,参考值10.00~60.00 μmol/L)。4岁2个月于南京医科大学附属妇产医院遗传医学中心就诊。体格检查:体质量12 kg,身高90 cm,不会说话,头围明显增大,四肢肌张力低,不能抬头,不能站立,不能抓物。患儿出生时,父亲24岁,母亲25岁,均体健,否认近亲结婚,否认遗传病家族史。患儿母亲于南京医科大学附属妇产医院就诊时已停经9周,确认早期妊娠。
为进一步对患儿进行明确诊断及治疗对患儿及其父母进行基因检测,所有检测前均经父母签署知情同意书,且经医院伦理委员会审核通过。采集患儿及其双亲外周血各2 mL,置抗凝管中-20℃冻存。取K 2 EDTA 抗凝外周血标本0.2 mL,用全血DNA 提取试剂盒(美国Omega 公司)提取DNA。采用Ion AmpliSeqTM Custom Panel(Life Technologies)对目标基因进行扩增。采用Ion Ampliseq Library Kit 2.0 和 Ion Xpress Barcode Adapter 1-16 Kit(Life Technologies)进行文库构建,对PCR 扩增产物加上序列标签及测序接头。用Ampure Beads Kit 对产物进行纯化,用Agilent2100 Bioanalyzer平台进行文库质控,制备的文库平均片段大小为300 bp 左右;采用Ion OneTouchTM System 和Ion PGMTM Hi-QTM View OT2 Kit(Life Technologies)进行乳液PCR,在Ion OneTouch ES系统(Life Technologies)上对模板阳性的磁珠颗粒(Ion Sphere Particles,ISPs)进行富集;采用Ion PGMTM Hi-QTM View Sequencing Kit 和Ion 318TM Chip V2(Life Technologies)在Ion Torrent PGM 测序平台上进行测序;采用Ion Torrent Suite V4.2软件进行Ion Torrent数据提取、序列比对及单核苷酸位点变异(SNVs)和Indels提取,得到的SNVs和indels经dbSNP 142数据库和千人基因组数据库过滤后,检索HGMD、LOVD 等数据库及PubMed 相关文献,进行变异位点致病性分析。对原始BAM 文件应用Integrative Genomics Viewer(IGV)软件进行比对读序(reads)的可视化分析。结果发现,患儿GCDH基因c.395 G>A(p.R132Q)、c.769C>T(p.R257W)复合杂合型突变(图1)。GCDH基因第6 外显子编码区c.395 G>A(p.R132Q)错义突变HGMD数据库ID为“CM000391”,SFIT软件预测为有害;GCDH基因第8外显子编码区c.769C>T(p.R257W)错义突变HGMD数据库ID为“CM980863”,SFIT软件预测为有害。
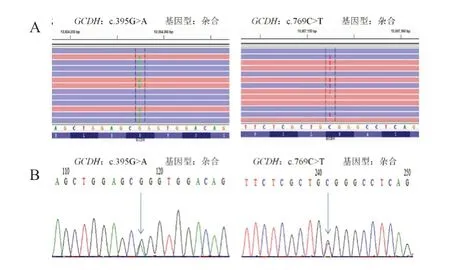
图1 患儿GCDH 基因测序结果
根据患儿变异位点,设计2 对P C R 引物。①GCDH-e6F:TCC TTA TTC AGC CCT GTC TCT T,GCDH-e6R:GCG TTC ATC GTC CAC TTC AA;②GCDH-e8F:CCA CTA CAA CTC ATC CAA CAA GA,GCDH-e8R:AGC GAG ACG GTC ACC AAA T。对GCDH基因第6、8 外显子进行PCR 扩增,琼脂糖凝胶电泳纯化后,用ABI 3130基因分析仪进行测序。结果用Lasergene 软件包中的SeqBuilder 程序进行序列比对,GCDH基因标准参照序列为NM_000159.3。经Sanger 测序验证发现,患儿母亲携带c.769 C>T(p.R 257 W),基因型为杂合;患儿父亲携带c.395 G>A(p.R 132 Q),基因型为杂合(图2)。患儿GCDH基因同时携带2 个已明确报道过的致病性突变(分别遗传自父亲、母亲),为复合杂合型突变,该基因型改变符合患儿临床表型及血液学检测结果,因此患儿明确诊断为戊二酸血症I型。
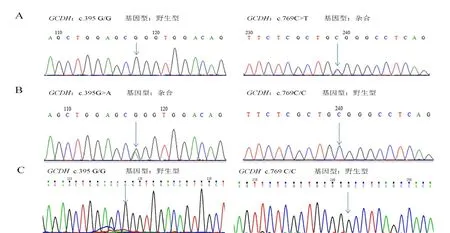
图2 患儿父母及胎儿GCDH 基因测序结果
由于患儿母亲已再次怀孕,经医学伦理审核及家属知情同意,于孕18 周进行羊膜腔穿刺,抽取羊水标本,提取DNA,进行产前基因检测。常规经腹羊膜腔穿刺抽取羊水30 mL,其中20 mL 行羊水培养,10 mL 行羊水标本母体污染检测及基因检测。羊水离心后用1 mL PBS液洗涤2次,细胞沉淀用Lab-Aid 820核酸提取仪(Lab-Aid核酸分离试剂盒,厦门致善公司)提取DNA 样本。采用21 三体和性染色体多倍体检测试剂盒(广州达安基因公司)对胎儿羊水细胞基因组DNA 及其父母外周血基因组DNA 进行扩增,对7个短串联重复序列(STR)位点进行分析,以排除母体DNA 的污染。7 个STR 位点包括6 对STR 位点(分别为D21S1435、D21S11、D21S1411、DXS981、DXS 6809、X 22)与1 个性别特异性(AMXY)位点。STR 检测结果表明未见母体污染。对羊水标本进行Sanger测序检测,结果显示,胎儿GCDH基因c.395位点、c.769位点均未发生突变(图2)。胎儿足月正常分娩,出生后取新生儿足根血干血片、尿液行血LC/MS及尿GC/MS检测,结果显示,新生儿戊二酰肉碱浓度、游离肉碱浓度、戊二酸浓度均在正常范围。电话回访,目前10 月龄,当地医院体检显示生长发育情况良好,无异常症状。
2 讨论
GCDH 是黄素蛋白家族中的一种酰基辅酶A 脱氢酶(acyl-CoA dehydrogenase,ACD),除了催化底物脱氢以外,它还可以催化底物乙酰辅酶A 脱羧基[3]。GCDH 的活性形式是由4 个单体构成的同源四聚体,每个单体是由438 个氨基酸组成的前体蛋白,其中44 个N-末端氨基酸是线粒体定位信号,在进入线粒体后会被切除。GCDH 单体的二级结构分成3 个结构域:N-末端α-螺旋(R 45~Q 167)、C-末端α-螺旋(S 282~K 438)及中间的β-片层(L 168~S 281),N-末端、C-末端α-螺旋都和右侧配体形成交互界面,中间的β-片层和左侧的配体相互作用[7]。在GCDH四聚体中,每个单体的中间β-片层、C-末端α-螺旋及邻近单体的C-末端α-螺旋形成一个空腔,空腔中通过非共价键结合1 分子黄素腺嘌呤核苷(flavin adenine dinucleotide,FAD),从而形成一个有催化活性的GCDH四聚体[8]。在线粒体基质中,同源四聚体GCDH 直接与其他线粒体基质蛋白接触形成多酶复合体,例如二氢硫S-琥珀酰转移酶(dihydrolipoamide S-succinyltransferase,DLST)、含有异源二聚体电子转移黄素蛋白FAD,协同完成催化作用。多酶复合体中DLST和GCDH的连贯催化作用是戊二酰辅酶A氧化脱羧基反应及维持细胞内氨基酸稳定的先决条件[9]。任何影响GCDH同源四聚体、多酶复合体结构形成的氨基酸残基改变均可能导致GCDH 催化活性降低或降解加速,使得细胞内相应底物堆积,最终导致机体出现与神经系统损伤相关的临床表型。在HGMD(The Human Gene Mutation Database)公共版数据库中,目前共有135 种可导致氨基酸改变的错义/无义点突变(截至2018 年1 月,专业版HGMD 数据共有171 种错义/无义点突变),其中38种氨基酸改变是位于GCDH单体N-末端α-螺旋结构域(R45~Q167),35种位于中间的β-片层(L168~S281)结构域,62种位于C-末端α-螺旋结构域(S282~K438),这种C末端突变明显占多的分布模式可能与C-末端α-螺旋结构域是GCDH主要的催化活性中心有关。
本例患儿为GCDH c.395 C>T(p.R 132 Q)、c.769 C>T(p.R 257 W)复合杂合突变,这2 个突变均为已报道过的致病突变[10-11]。R 132 位于GCDH 单体N-末端α-螺旋结构域D 区段,R 257 位于GCDH 单体中间β-片层结构域6、7 区段中间。R(精氨酸)是极性正电荷亲水氨基酸;Q(谷氨酰胺)是极性不带电荷氨基酸,亲水性明显低于R;W(色氨酸)是非极性疏水氨基酸。已有研究者通过软件分析GCDH 的3 D结构,显示p.R 132 Q 可影响GCDH 单体四聚化[12]。在氨基酸组成、折叠成特定蛋白质二级结构的过程中,带电荷亲水氨基酸多位于立体结构的表面,疏水氨基酸多位于内部。p.R132Q、p.R257W突变可能因此改变了GCDH单体的二级结构折叠,进而阻碍了同源四聚体的形成。同时,p.R 132 Q、p.R 257 W 突变可能导致GCDH单体蛋白分子表面的静电点位改变,使得GCDH 单体四聚化过程及多酶复合体的聚合过程受阻。以上分子机制推断需要进行定点突变后的蛋白晶体结构分析、蛋白生化活性检测等实验手段来验证。受实验室的条件所限,本例患儿未能行以上验证实验。
GA I是一种可治疗的疾病,但是在出现严重不可逆的中枢神经系统症状之前,患儿不会表现出特异性症状,这使得早期临床诊断很困难。目前,许多国家和地区已将GA I 加入到新生儿筛查项目中,通过串联质谱技术结合GCDH基因突变检测方案可极大地提高诊断的敏感性和特异性[13-14]。同时,对于有再生育要求的患儿家庭来说,GCDH基因突变分子遗传学检测技术使得产前诊断成为可能。本研究成功确定了GD I 家系GCDH基因致病突变位点,并为患儿母亲再生育实施了产前分子遗传学诊断,确保新生婴儿的健康。
综上,分子遗传学检测是GA I有效的确诊方法,应用串联质谱筛查技术、高通量测序技术及产前诊断相结合的联合诊疗方案可有效的减少GA I患儿的出生。
猜你喜欢
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
广州大学学报(自然科学版)(2019年1期)2019-05-07 01:33:26
生物学通报(2019年3期)2019-02-17 18:03:58
中国军转民(2017年7期)2017-12-19 13:30:00
天津科技大学学报(2016年1期)2016-02-28 16:59:45
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10 08:41:53
大连工业大学学报(2015年4期)2015-12-11 04:06:50
现代检验医学杂志(2015年2期)2015-02-06 02:01:01
中国卫生(2014年10期)2014-11-12 13:10:24
