
德朗热综合征1例基因检测和临床分析
2020-04-04 10:50胡宇慧陈淑丽
临床儿科杂志 2020年3期
胡宇慧 陈淑丽 刘 麟 崔 冬
深圳市儿童医院 1.遗传代谢科,2.心血管内科(广东深圳 518038)
德朗热综合征(Cornelia de Lange syndrome,CdLS,MIM:122470,300590,300882,610759,614701)又称为阿姆斯特丹侏儒症,是一种罕见的多系统受累的发育障碍性遗传疾病,具有独特颅面特征、生长迟缓、认知障碍、肢体缺陷、多毛症和多系统受累等临床特点[1]。独特的面部特征为CdLS 的主要诊断点,包括弓形眉、连眉、多毛、短鼻、鼻孔上翘、薄上唇和小下颌等。CDLS 的发病率占活产婴儿1/200000~1/10000[2]。目前明确为CdLS致病基因有5个:NIPBL、SMC1A、SMC3、RAD21和HDAC8。有报道提示,BRD 4和ANKRD 11基因与CdLS 相关[3]。遗传方式为常染色体显性遗传(NIPBL、SMC3、RAD21)和X-连锁显性遗传(SMC 1 A和HDAC 8),多数为散发病例,<1%的患者有同样受累的父母[4]。参考人类基因突变数据库(Human Gene Mutation Database,HGMD),NIPBL为CdLS最常见致病基因,至今报道约300个突变位点,无热点突变报道。目前CdLS无特效治疗方法。本文通过对1 例高度疑似CdLS 患儿的全外显子组基因突变检测,报道一个新发现的NIPBL基因变异位点,同时进行临床资料总结,以便增加临床医师的认识。
1 临床资料
患儿,男,6 月龄,因体质量增长缓慢6 个月就诊于遗传代谢科。患儿G3P2,孕37+4周剖宫产出生,出生体质量2.1 kg(<-3 SD),身长43 cm(<-3 SD),Apgar评分不详,无窒息抢救病史。生后因面容特殊在当地住院半个月,诊断为“先天性脑发育异常,先天性心脏病(肺动脉狭窄),新生儿肺炎,足月小样儿,隐睾”。患儿曾祖父母为表兄妹,父母体健。母亲孕期多次彩色超声示胎儿宫内生长受限,拒绝行羊水穿刺,曾住院保胎治疗,既往人流1 次。患儿7 岁姐姐体健。患儿出院后至今混合喂养,平素进食偏慢,无频繁呕吐,无慢性腹泻,无反复感染病史,无抽搐,体质量增长缓慢,目前会无意识笑,抬头不稳,哭声响亮,听力视力无明显异常。体格检查:体质量3.3 kg(<-3SD),身长55 cm(<-3SD),头围32.5 cm(<-3SD)。精神尚可,前囟2 cm×2 cm,额部多毛,连心眉,弓形浓眉,睫毛长且弯曲,双眼睑下垂,鼻梁低平,人中长,嘴唇薄,嘴角下斜(图1)。心前区可闻及3/6级收缩期杂音。胸廓无畸形,肺腹无异常;左侧隐睾;双手小,左手通贯掌,双足第四趾短。四肢肌力尚可,肌张力无异常。实验室检查:近期外院血常规、胸片、肝酶、心肌酶谱、电解质、甲状腺功能均无异常,血串联质谱和尿气相色谱质谱结果阴性。染色体微阵列阴性。彩色多普勒超声心动图示肺动脉狭窄。生殖器超声示双侧睾丸下降不全。
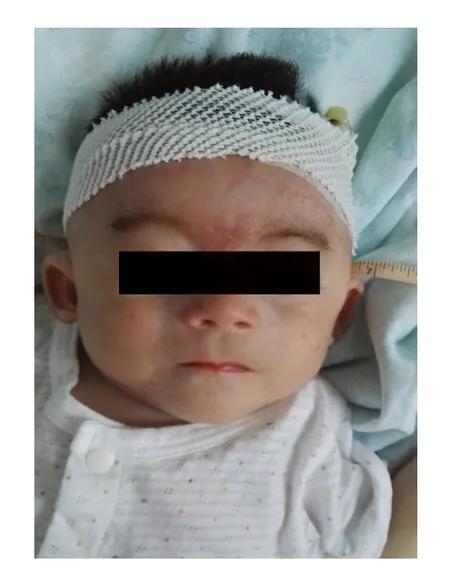
图1 患儿面部特征
经医院伦理委员会审核及患儿父母知情同意,EDTA 抗凝管采集患儿和父母外周血各2 mL 行基因分析。采用吸附柱法(QIAGEN公司)提取纯化全基因组DNA;采用超声波方法(Covaris公司)对DNA片段化并通过低循环数PCR将其两端加上index标签及测序元件,经全外显子V6版探针(Agilnet公司)进行目标区域捕获并洗脱纯化得到测序前文库样品。制备好的文库通过NovaSeq(Illumina公司)二代高通量测序平台进行测序。测序完成后利用软件对fastq数据进行过滤并使用BWA、比对、变异识别等生物信息分析,结合外部公共数据库、临床表型信息,参照美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)进行变异解读。利用Sanger 测序法对患儿及父母进行阳性及疑似突变位点验证(深圳基因启示录公司完成)。引物设计采用Primer5.0软件,相关序列见表1。

表1 NIPBL基因第9外显子扩增上下游引物序列
检测结果发现,患儿NIPBL基因第9外显子存在NM-133433 c.1679delA(p.K566Sfs*48)杂合突变,该突变导致突变位点后48 位出现终止密码子,转录蛋白出现截短,应用Sanger测序对患儿突变位点进行家系验证,证实患儿为杂合子,患儿父母该基因位点均无异常。见图2。根据ACMG 联合美国分子病理学会(Association for Molecular Pathology,AMP)2015年制订的“基因序列变异的解释标准和指南”进行致病性分析[5],c.1679delA导致缺失突变后48个异常氨基酸后出现截短蛋白,是无功能突变,为致病突变(非常强致病性证据,PVS 1)。通过比照千人基因组数据库(1000 Genomes)、人类基因突变数据库(HGMD)和Clinvar数据库及近5年文献,未见c.1679delA突变报道和收录(中等致病性证据,PM2);c.1679delA为未经父母验证的新发变异(中等致病性证据,PM 6);患儿的临床表现与NIPBL基因突变导致的CdLS 表型高度吻合(支持性致病性证据,PP 4)。因此根据ACMG 评级规则,c.1679 delA 突变(PVS 1+PM 2+PM6+PP4)判断为未见文献报道的致病性变异。
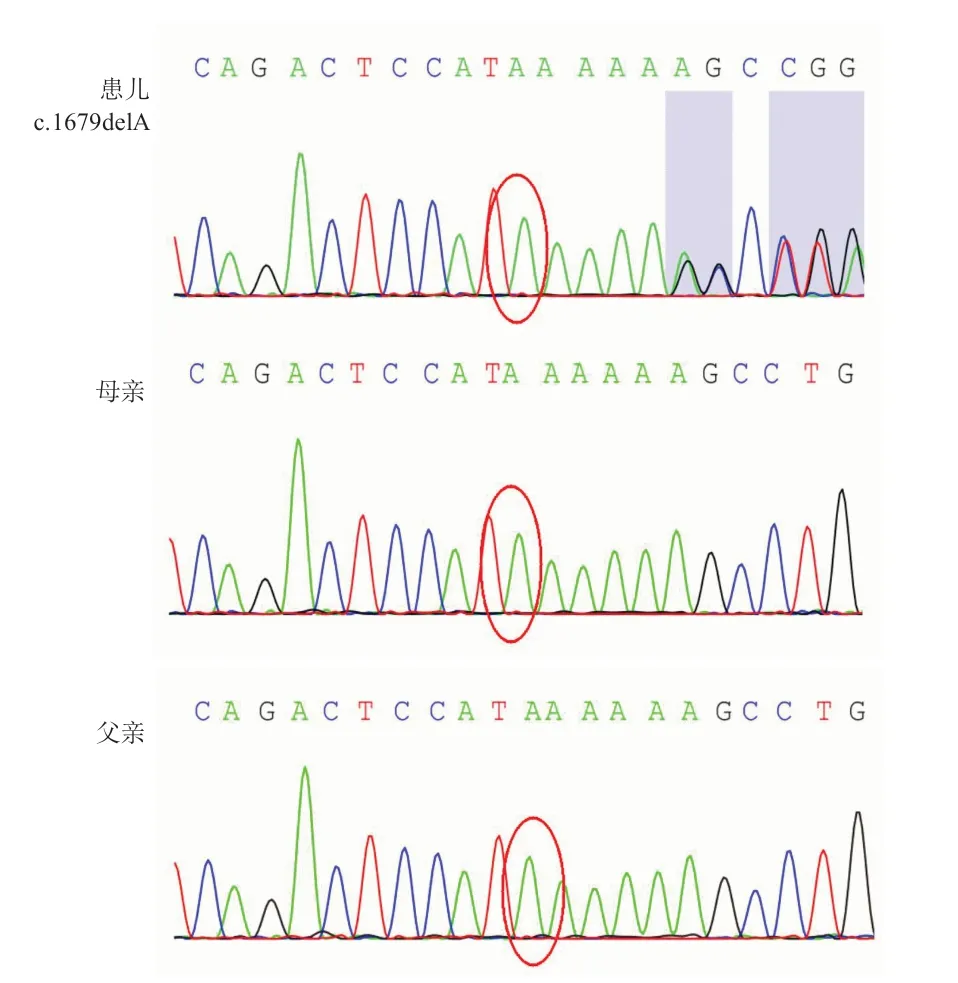
图2 患儿及父母NIPBL 基因突变测序结果
2 讨论
CdLS于1933年首先由荷兰籍儿科医师在2例发育障碍患儿中描述。2018 年发表针对CdLS 的诊断和处理的专家共识[6]。共识评分标准:①主要特征(每一条记2分):连眉和/或浓眉;短鼻,鼻梁塌陷和或鼻孔前倾;长和/或平人中;上嘴唇薄和/或嘴角下弯;手少指和/或无指;先天性膈疝。②次要特征(每一条记1分):全面发育迟缓和或智力障碍;宫内生长迟缓(<2 SD);生后生长迟缓(<2 SD);小头畸形(宫内和或生后);小手和或小脚;小指短;多毛。根据专家共识,CdLS诊断标准为评分≥11分,即使没有分子遗传学检测也可临床诊断为CdLS。本例患儿根据评分标准的临床评分为13 分,可诊断为经典型CdLS。有多个基因(EP300、AFF4、NAA10)缺陷临床表现与CdLS临床表型有部分重叠,为类德朗热样综合征。CdLS基因检测中约15%~20%出现嵌合现象[7],应用一代测序方法检测外周血淋巴细胞可能出现漏诊,此外约30%临床诊断为CdLS患者的遗传背景尚不清楚。由于CdLS 诊断经验不足,本例患儿选择全外显子组基因检测技术对外周血进行分子诊断学研究。
Cohesin 复合物主要功能为参与姐妹染色单体聚合,确保有丝分裂和减数分裂过程中染色体的正确分离,同时在DNA修复、细胞功能维持和分化以及基因表达调控中具有非常重要的作用[8]。Cohesin复合物相关蛋白编码基因发生突变导致CdLS的发生。SMC1A、SMC3和RAD21编码cohesin复合物的三个核心亚基,NIPBL和HDAC8编码蛋白为cohesin复合物调节蛋白。在明确与CdLS相关的5个基因中可找到约70%的致病突变,NIPBL为CdLS最常见致病基因,在具有典型临床表型的CdLS中NIPBL致病突变检出率为60%[9]。在对基因型与临床表型的相关性研究中表明,携带NIPBL突变患者与SMC1A、SMC3、RAD21和HDAC8突变的患者相比更易出现严重表型[10]。NIPBL中具有无义、剪接位点和移码突变导致截短突变的患者比具有错义突变的患者具有更严重的表型[11]。本例患儿在NIPBL基因上检测到一个缺失突变,导致缺失突变后48个异常氨基酸出现截短蛋白,临床表现为经典型CdLS,与文献报道一致。NIPBL外显子32缺失会影响复合物中NIPBL编码的蛋白与其他蛋白结合,临床表型严重[3],c.1679delA 突变导致第9外显子后包含外显子32编码的蛋白质缺失,可能为该突变临床表型严重的原因之一。本例患儿父母未检测到相应变异,临床表型正常,考虑患儿为自发突变可能。有报道一对正常夫妇由于性腺嵌合,孕育两次NIPBL突变胎儿[12]。因此不能排除患儿父母生殖细胞嵌合体可能,在父母孕育下一胎时建议羊水或绒毛膜穿刺进行产前诊断。此外,有报道CdLS 基因检测中约15%~20%出现嵌合现象,外周血检测阴性,口腔颊黏膜细胞或者皮肤成纤维细胞检测阳性[7,13]。本例患儿父母仅进行外周血检测,不能排除为嵌合体可能,但患儿父母无特殊面容,智力正常,可能性较小。
CdLS 为类似于唐氏综合征和脆性X 综合征一样具有典型的畸形特征和智力迟钝的一种疾病。文献报道,连眉、浓眉、多毛和喂养困难见于90%以上CdLS患者[14]。本例患儿孕期就存在宫内发育受限,出生后发现面容特殊、体质量增长缓慢。有文献报道,80%CdLS患儿在妊娠中期发现对称性宫内生长受限[15]。对于没有明确基因突变类型的下一胎产前诊断可行B 超了解胎儿生长发育及面部轮廓、肢体异常来诊断。本例患儿在孕期发现宫内发育迟缓,进行产前分子遗传学检查可能有助于减轻家庭的负担[4]。在肢体异常中,上肢较下肢畸形更常见及严重,上肢畸形中可出现小手、第一掌骨缩短、短指、并指、近端桡骨头发育不全、尺骨缺损等,下肢畸形通常较轻微,包括小脚,缩短的趾骨和双侧马蹄足等,NIPBL中截短蛋白突变更易出现严重型肢体畸形[15]。本例患儿为NIPBL截短蛋白突变,肢体缺陷仅表现为小手和第四趾骨缩短,但未行双上肢的X线检测,不能完全排除存在近端桡骨头发育不全和尺骨缺损可能。
本例患儿以足月小样儿及生后生长缓慢为主要临床表现,伴有特殊面容和多发畸形,临床医师易倾向于染色体组病,尽管CdLS 也有少数大片段缺失报道,但主要为点突变。本例患儿在外院行染色体微阵列检测未发现异常,因此临床在诊治类似患儿时应首选基因检测,减少患儿家长经济负担。由于CdLS存在体细胞嵌合现象,外周血淋巴细胞检测可出现阴性,建议口腔颊黏膜细胞为首选检测标本。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
心电与循环(2021年4期)2021-11-29
中国生殖健康(2020年4期)2021-01-18
中学数学杂志(2019年9期)2019-05-29
中国生殖健康(2018年4期)2018-11-06
现代园艺(2017年21期)2018-01-03
中国眼镜科技杂志(2016年17期)2016-10-24
中国康复理论与实践(2015年10期)2015-12-24
医学研究杂志(2015年5期)2015-06-10
现代检验医学杂志(2015年5期)2015-02-06
