
KCNA2 基因致早发性癫痫性脑病 1 例临床和基因变异分析
2020-04-04 10:50王卫卫崔清洋何晓敬
临床儿科杂志 2020年3期
王卫卫 崔清洋 何晓敬
新乡医学院第一附属医院儿科(河南卫辉 453100)
早发性癫痫性脑病(early epileptic encephalopathy)是一类由于难治性癫痫发作及严重异常脑电导致的严重脑疾病,主要特征为新生儿或婴儿早期起病,难治性的癫痫发作及严重的脑电图异常放电,精神运动发育迟滞或倒退[1]。根据发病年龄、发作类型及脑电图等的特点,2010 年的国际抗癫痫联盟对早发性癫痫性脑病进行了命名及归类,主要包括新生儿期发病的早发肌阵挛脑病、大田原综合征,婴儿早期发病的婴儿恶性游走性部分性发作、West 综合征和Dravet 综合征,以及维生素依赖性脑病等[1]。KCNA2基因变异所致的早发性癫痫性脑病遗传方式为常染色体显性遗传,纯合变异或杂合变异均可致病。目前关于KCNA2基因变异所致早发性癫痫性脑病的发病率尚不清楚,临床报道均为个案或小队列研究,其所引起的癫痫发作为难治性,预后差。本文回顾分析1例国内首次报道的由KCNA2基因变异所致的早发性癫痫性脑病患儿的临床资料及基因检测结果。
1 临床资料
女性患儿,42+6周出生,出生后30 分钟即出现间断抽搐,于出生3 小时入院。患儿为G2P1,足月,因过期产、胎儿宫内窘迫行剖宫产,出生1分钟及5分钟Apgar评分分别为9分、10分。父母体健,非近亲结婚。患儿的舅舅6岁时罹患化脓性脑膜炎后出现继发性癫痫。入院体格检查:一般情况较差,反应差,面色稍发绀;全身皮肤黏膜及肢端稍发绀;头颅大小正常,无畸形,无血肿及产瘤,前囟稍饱满,张力稍高,心肺腹无异常;四肢肌力无异常,肌张力稍高,原始反射未引出。实验室检查:血常规、肝肾功能、心肌酶、电解质、超敏C反应蛋白、降钙素原、血浆氨及乳酸均无异常。入院后腰椎穿刺脑脊液压力、常规及生化检查均无异常。头颅 CT 和磁共振成像(MRI)未见明显异常。24小时动态脑电图示发作期左侧中央、中颞部为著的尖波发放。患儿入院时抽搐呈局灶性。入院后予止惊、静脉营养、预防出血等治疗,住院期间抽搐频繁,并发展为全身性抽搐,曾多次调整抗癫痫药物(苯巴比妥、水合氯醛、地西泮、丙戊酸钠、左乙拉西坦、氯硝西泮、奥卡西平)仍无效,住院 58 天后患儿仍有全身强直发作,家属自动出院。
经医院医学伦理审核,家长知情同意后,取患儿外周血及尿液送北京敏路思检验所行血串联质谱及尿气相色谱质谱检验;同时抽取患儿及父母外周血各 2 mL 送北京康旭医学检验所行相关基因检查,引物序列及 PCR 片段大小见表 1。血串联质谱及尿有机酸检测提示患儿辛二酰甘氨酸-2 增高,但不足以解释其临床表型。二代测序并 Sanger 测序验证显示,患儿c.1120A>G(编码区第 1120 号核苷酸由 A 变为G)的杂合核苷酸变异,该变异导致第 374 号氨基酸由 苏氨酸变为丙氨酸(p.Thr 374 Ala),为错义变异。该变异可能导致蛋白质功能受到影响。患儿父母该位点均未见异常(图1)。该变异经 SIFT 和 Polyphen2及 Mutation Taster 软件预测均可能有致病性,且该基因位点的致病性国外已有文献报道[2]。该变异不属于多态性变化,在人群中发生的频率极低(所参考数据库:1000Genomes、dbSNP)。KCNA2基因所致早发性癫痫性脑病为常染色体显性遗传,杂合变异可能导致发病。患儿父母KCNA 2基因均未发现上述变异,是为新生变异,此变异可能为致病性变异。患儿确诊为KCNA2基因c.1120A>G 变异所致早发性癫痫性脑病。但抗癫痫治疗效果差,最终家属放弃治疗,出院20天后死亡。
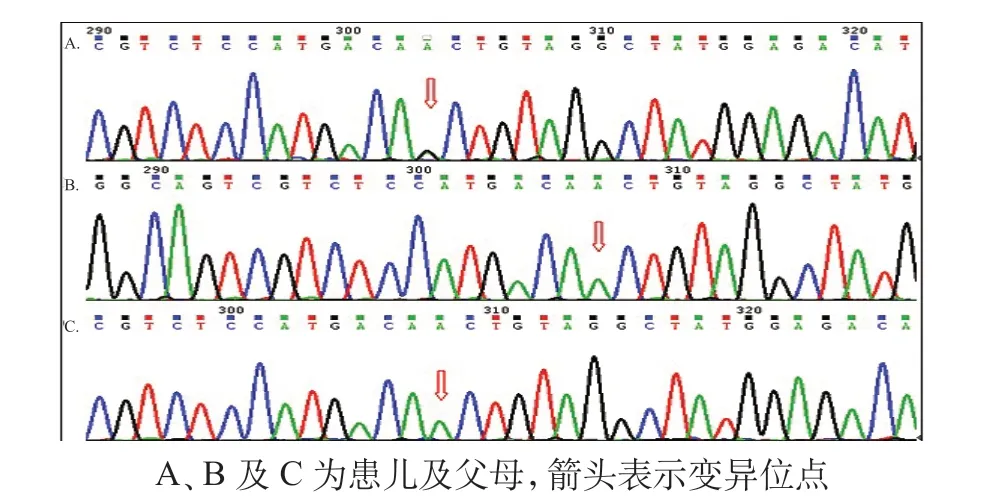
图1 KCNA2 基因测序峰图
对KCNA2基因进行蛋白质二级结构预测显示,该基因包括152 个α-螺旋,0个β-折叠,120个延伸链和227个无规卷曲。其中二级结构所占比为:α-螺旋为 30.46%,β-折叠为 0%,延伸链为 24.05%,无规卷曲为 45.49%。c.1120A>G(p.Thr374Ala)突变位于无规卷曲使原来的极性中性氨基酸苏氨酸变为非极性疏水性氨基酸丙氨酸,为错义变异,位于蛋白质编码区,推测可能会导致功能上的一些改变。
2 讨论
近年来,遗传因素在癫痫发病机制中作用的研究日渐受到重视。至今共发现 977 个基因与癫痫相关,其中 60 个是离子通道基因[3]。癫痫的遗传方式多样,常染色体显性遗传最为常见。离子通道基因在癫痫的发病中起到了非常重要的作用。
与癫痫相关的离子通道基因突变主要包括电压门控离子通道、神经元递质受体门控通道基因突变及其他少数编码非离子通道蛋白的基因突变。近年来研究发现,编码离子通道基因突变与众多原发性癫痫的发病高度相关。其中钾离子通道是调节细胞膜电流变化的最大离子通道蛋白族,主要功能是维持细胞膜的静息电位,并通过介导钾离子外流使细胞膜存在复极化,从而控制细胞的兴奋性。由于电压门控钾通道在中枢神经系统中的表达,在神经兴奋性和神经递质释放中发挥重要作用,KCNA2基因是癫痫性脑病发生发展的一个强有力的候选基因,已有报道该基因的突变导致神经性疾病。
KCNA2基因编码Kv1.2通道,该通道主要存在于轴突和突触末端,能够保障细胞膜在动作电位后产生有效的复极化。研究证实,KCNA2基因的5个错义突变与癫痫性脑病相关[4-5]。而在动物模型的研究中发现,KCNA2敲除小鼠较正常小鼠更易发癫痫且存在过早死亡[6],从而更加证实了KCNA2在癫痫发病中的重要作用。

表1 KCNA2基因引物及 PCR 扩增反应条件
编码 Kv 1.2 通道的KCNA 2基因定位于染色体1p13.3,全长约3 100 bp,含3个外显子及2个内含子,编码499 个氨基酸。Kv 1.2 通道由具有6 个跨膜片段(S1-S6)的4个亚单位组成,跨膜片段 S1-S4 形成电压传感器域,S5-S6 形成孔区域,其中包含选择性过滤器和门控离子流[7]。有报道在23例患者中共检出两种变异类型,22例的10 种错义变异和1例的截短变异,且20例为新生变异[7]。
本例患儿检测到CNA2基因c.11 2 0 A >C(p.Thr374Ala)错义变异,父母均未检出,为新生变异,符合文献常见变异类型,且该基因位点的致病性已有报道[2]。同时分析c.1120A>G位点位于高度保守区域(变异位点保守值为5.71,>2 即认为是保守区域,变异位点值参考数据库为 GERP++RS 数据库),且位于重要蛋白结构区域。对KCNA 2基因进行蛋白质二级结构预测显示,该基因包括 152 个α-螺旋,0 个β-折叠,120 个延伸链和 227 个无规卷曲。其中二级结构所占比为α-螺旋30.46%,β-折叠 0%,延伸链24.05%,无规卷曲 45.49%,c.1120A>G(p.Thr374Ala)变异位于无规卷曲,使得原来极性中性氨基酸苏氨酸变为非极性疏水性氨基酸丙氨酸,为错义变异,位于蛋白质编码区,推测并结合文献该错义变异不会产生任何功能蛋白[2]。
迄今为止,对4种致病性KCNA2变异的功能研究表明,其可导致显性负效的功能缺失或显著的功能获得[4],且依据脑病和癫痫的严重程度分为两组,即临床症状较轻的功能缺失变异组和临床症状较重的功能获得变异组[8]。Kv 1.2 属于延迟整流类的钾通道,使神经元在动作电位后能够有效地复极化。功能缺失变异预测过度兴奋的神经细胞膜和由于复极化受损而致重复的神经元放电,KCNA2基因敲除小鼠的癫痫表型证实了这一假设[6]。与之形成鲜明对比的是,R297Q和 L298F(均为功能获得变异)预测生理性膜电位处于永久性开放通道以及膜超极化引起的电沉默。
研究报道,8例功能缺失变异患儿,癫痫发作中位年龄为 8.4 个月(范围2~17 个月),其中3例表现为热性惊厥(1例持续性热性惊厥),6例为局灶性癫痫发作(4例为偏身强直发作,且其中2 例先表现为眼球偏斜和呕吐);中位随访 4 年(范围1.5~6 年),4 例无癫痫发作,1例很少继续发作[8]。而另外功能获得变异9例患儿癫痫发作的中位年龄为 8.7 个月(5~15 个月),其中 1 例自出生时即发作四肢和头部伸展或屈曲。在发病过程中,所有患儿均表现为全身性癫痫发作,且有 8 例患儿癫痫发作未控制[8]。
本例患儿出生后不久即癫痫发作,且发作频繁,多种抗癫痫药物治疗效果差,结合文献及软件预测考虑为功能获得变异。
本文报道1例KCNA2基因c.1120A>G 变异致早发性癫痫性脑病病例,为国内首次报道,扩充了国内早发性癫痫性脑病的基因突变谱。
猜你喜欢
广西医科大学学报(2022年5期)2022-06-07
世界科学技术-中医药现代化(2022年2期)2022-05-25
昆明医科大学学报(2022年3期)2022-04-19
中老年保健(2021年12期)2021-08-24
皮肤病与性病(2021年3期)2021-07-30
世界科学技术-中医药现代化(2021年12期)2021-04-19
当代水产(2020年4期)2020-06-16
中南医学科学杂志(2019年6期)2019-12-05
听力学及言语疾病杂志(2018年1期)2018-01-23
中国卫生标准管理(2015年17期)2016-01-20
