
肢带型肌营养不良4 家系临床和基因突变分析
2020-04-04 10:50邓胜勇田小会蒲向阳王言覃
临床儿科杂志 2020年3期
邓胜勇 田小会 姚 静 蒲向阳 王言覃 钟 敏,
1.重庆医科大学附属儿童医院神经疾病诊治中心 儿童发育疾病研究教育部重点实验室 儿童发育重大疾病国家国际科技合作基地 儿科学重庆市重点实验室 国家儿童健康与疾病临床医学研究中心(重庆 400014);2.重庆市黔江中心医院小儿呼吸内科(重庆 409099)
肢带型肌营养不良(limb-girdle muscular dystrophy,LGMD)是一组肌无力主要累及肢体近端为特点的遗传性肌营养不良,多数病例呈常染色体隐性或显性遗传。根据不同的致病基因分型命名,常染色体显性遗传的命名为LGMD 1 A、1 B、1 C 等,常染色体隐性遗传类型命名为LGMD2A、2B、2C等[1-5]。
国内报道的LGMD 以2 A 和2 B 为主,2 C 及2 E型LGMD 报道少。本文总结4 个家系,共5 例(3 例2 C 型,2 A 和2 E 型各1 例)确诊的LGMD 患儿的临床资料。
1 临床资料
回顾分析2015 年6 月—2018 年12 月在重庆医科大学附属儿童医院神经内科门诊诊断的肢带型肌营养不良患儿。共4个家系5例患者,男3例、女2例。本研究经重庆医科大学附属儿童医院科研伦理审查(2018-64)通过。
例1患儿是因发现血清肌酸激酶和转氨酶显著增高就诊,没有肌无力的临床表现。例4患儿因弟弟(例3)确诊后行基因检查确诊,有轻微肌无力。其余3 例患儿均有不同程度的肌力下降,部分有肌萎缩和/或腓肠肌假性肥大。患儿的主要临床特征见表1。
5例患儿及家属均采集外周静脉血各2 mL,提取基因组DNA,例1~例4在北京迈基诺医学检验所行神经肌肉病panel测序,对检测出的突变位点Sanger测序验证,家系成员行该位点Sanger测序验证。病例5在广州嘉检医学检验所,用二代测序平台测序,对发现的致病或可疑致病突变位点,进一步针对该基因进行高通量测序验证。所有突变均经生物信息学蛋白功能预测软件SIFT、PolyPhen_2、REVEL预测期致病性,查阅HGMD、Clinvar等数据库明确是否有该突变的致病性报道,并综合家系信息等分析,根据ACMG指南判定其致病性。基因检测获得患儿或监护人的知情同意。
基因检测结果,例1CAPN 3基因复合杂合突变,位于exon 4 的c.632+4 A>G 导致剪接突变,exon 5 的c.725dupG为移码突变(图1),导致后续氨基酸系列改变。有报道此2种突变系致病性突变[6-7],故该复合杂合变异系此患者的致病位点。例2 检测到SGCG基因纯合突变,c.768delC(缺失胞嘧啶),导致氨基酸改变p.S257Afs*23(移码突变)(图2)。蛋白功能预测结果为未知,疑似致病性变异。Clinvar数据库有此突变致病的报道。结合血生化结果、临床表现和家系特点,可判定该突变为致病突变。例3有SGCG基因c.320C>T纯合突变(图3),导致第107位丝氨酸错义突变为亮氨酸,蛋白功能预测结果为未知,疑似致病性变异。数据库中未见此突变致病报道,正常人群数据库频率为未知。但结合血生化结果、临床表现和家系特点,可判定该突变为致病突变。例4 行基因位点验证,亦有此致病性突变(图3)。例5有SGCB基因的纯合变异(c.1A>G,p.M1?),见图4,此突变可导致蛋白质翻译的起始密码子突变,预测会影响蛋白质翻译过程的启动。Clinvar数据库有此突变致病的报道。故判定系致病突变。
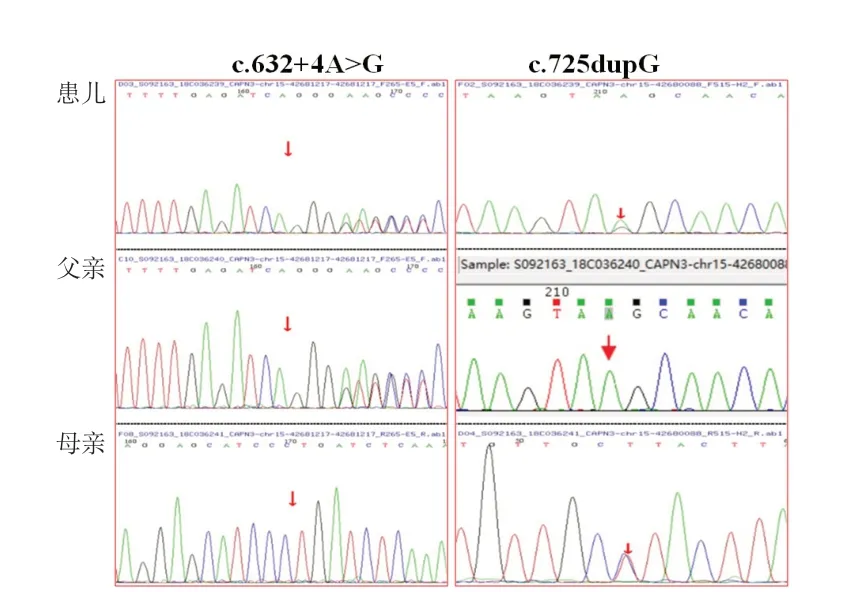
图1 例1 患儿及父母基因检测结果
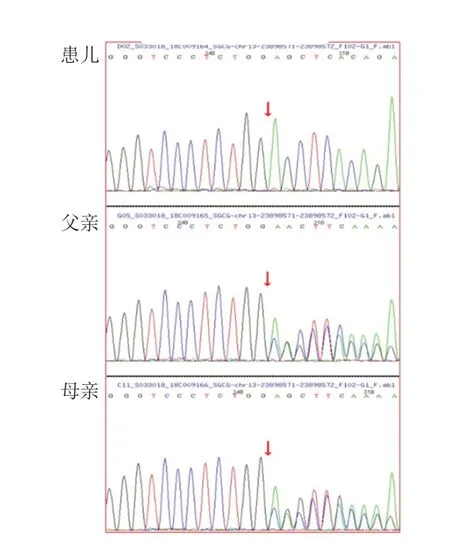
图2 例2 患儿及父母基因检测结果
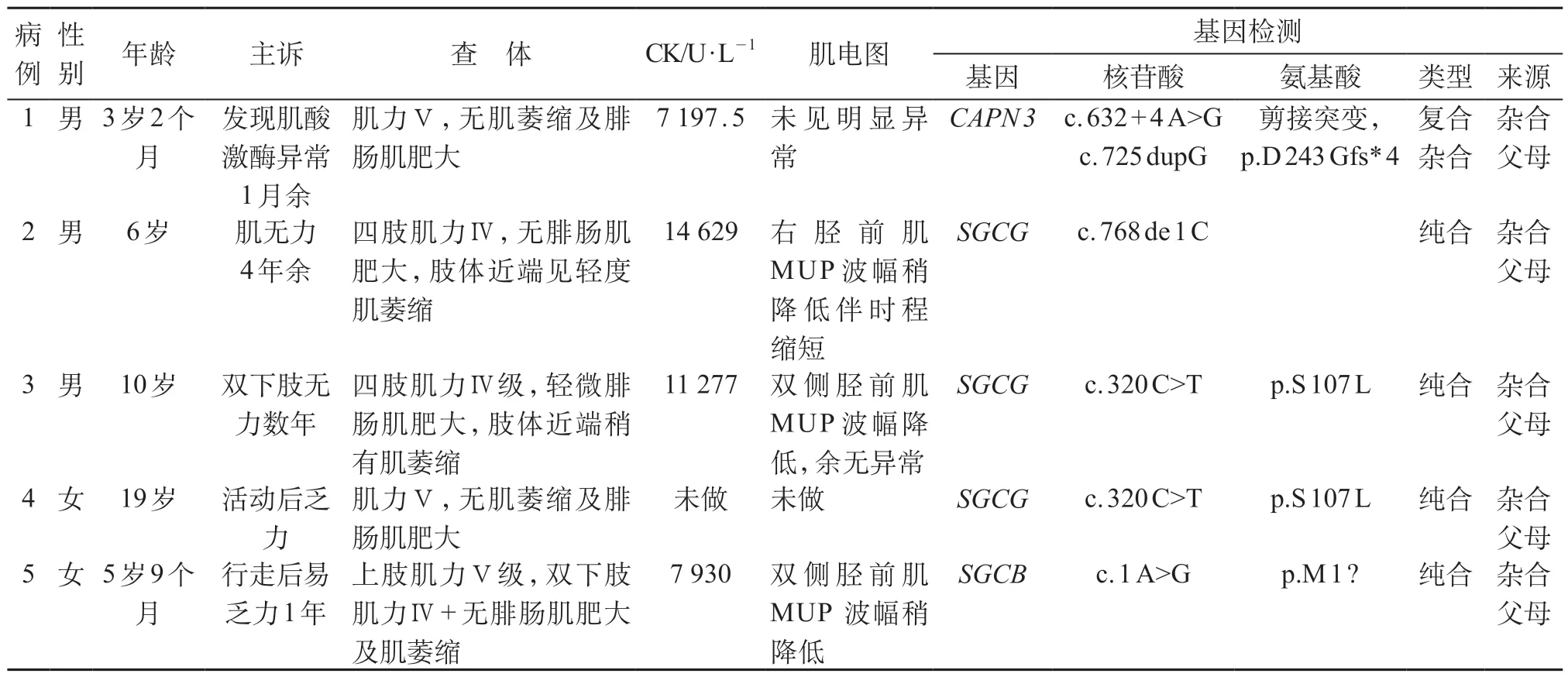
表1 5例肢带型肌营养不良儿临床资料和基因检查结果
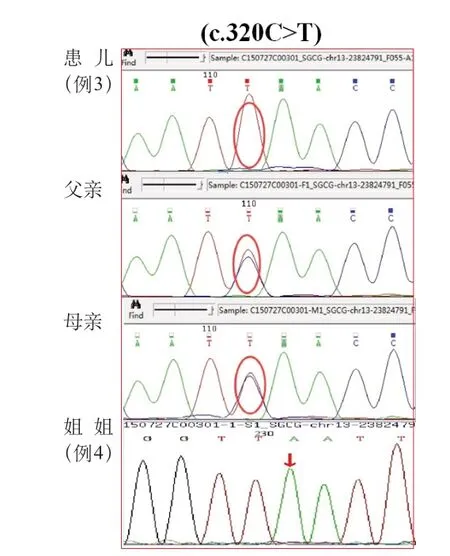
图3 例3 和例4 患儿及父母基因检测结果
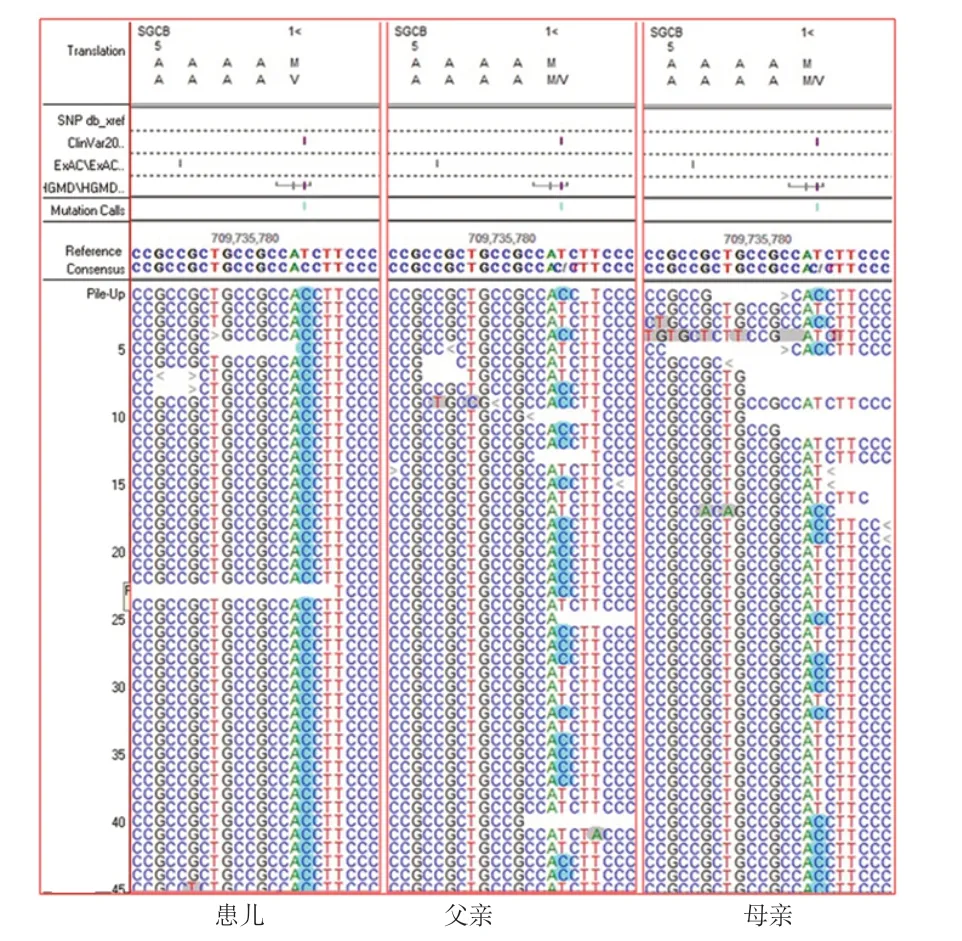
图4 例5 患儿及父母 基因检测结果
2 讨论
LGMD的肌无力主要累及肢体近端,如骨盆带和肩胛带肌无力,在疾病严重程度、表型、病理、发病年龄等方面具有临床异质性[1-5]。90%以上LGMD的病例系常染色体隐性遗传,其中以2A型最为常见,其次为2B型和2C型,而2E型极其少见[2]。
LGMD 2 A 是由编码蛋白水解酶钙蛋白酶-3 的基因(CAPN 3)突变引起,系常染色体隐性遗传,是目前最常见的LGDM 类型,占所有LGMD 病例的15%~40%[2,8-12]。71%的LGMD2A患者在6~18岁起病,主要累及肩胛周肌肉、肱二头肌、臀大肌、内收肌及腘绳肌,骨盆带肌无力临床表现较肩胛带肌无力重,严重的肌无力可影响髋关节及膝关节活动[13]。迄今为止报道的LGMD2A的CAPN3致病性突变有400多种。一项来自欧洲人群的研究显示,238例LGMD2A患者中,2362AG→TCATCT占所检测出CAPN3突变的30.7%[13]。智利的研究显示,218 例LGMD 2 型患者中,c.550delA突变占检测出的CAPN3基因总突变位点138个的47.1%[14]。来自意大利的研究显示,141例LGMD2A患者中,发现82种不同的CAPN3突变类型,其中12 例有550 delA,频率最高[15]。一项来自中国的资料显示,26例CAPN3突变的LGMD2A患者有6 例有c.2120 A>G,是占比最高的突变[6]。其他国家和地区的相关研究纳入例数较少,还难以判断相对的热点突变位点。本组患儿中,例1年龄小,尚未出现肌无力表现,但血清肌酸激酶显著升高,基因检测显示CAPN3基因复合杂合突变,其父母为无症状杂合携带者。此复合杂合突变位点均有报道系致病位点,但非热点突变,结合家系分析可确诊为LGMD2A型。
LGMD 2 C 和2 E 也都是相对常见的LGMD,均属于其中Sarcoglycan(肌聚糖)肌病的不同亚型。我国研究显示,Sarcoglycan 肌病在LGMD 中占11.5%~22.67%[9,16]。2 C 和2 E 型LGMD 临床表现相对类似,呈近端肌无力、翼状肩、腓肠肌肥大、巨舌及腰椎前凸。血清肌酸激酶常在500~20 000 U/L。LGMD2C致病基因SGCG位于13q12,其8号外显子突变导致编码的γ-肌聚糖功能异常而致病。目前共有70余种SGCG致病性突变,因为不同研究报道的例数都比较少,故尚未发现有特别的热点突变位点。本组患儿中,例2 有肌无力和肌电图改变,但未见典型的腓肠肌假性肥大表现,基因检查发现SGCG基因的cDNA 上768 号位置C 碱基(胞嘧啶)缺失,导致氨基酸水平上,第257 号位置由原本的丝氨酸(S)变成丙氨酸(A),并往后至第23 个氨基酸处发生终止,从而导致蛋白功能异常致病,故确诊为LGMD2C。例3有肢体乏力和肌酸激酶显著增高,伴有腓肠肌肥大和肢体肌萎缩。SGCG基因cDNA 上320 号位置C 碱基错义突变为T 碱基(胸腺嘧啶),导致第107 位置丝氨酸(S)变为亮氨酸(L),进而引起蛋白功能异常,故可确诊LGMD2C,此突变为未见报道的新的突变位点。例4系例3患儿的嫡亲姐姐,病史追问有肢体乏力,但可正常生活,因社会原因没能进一步完善肌酸激酶和肌电图等检查,故仅能疑诊LGMD2C。
LGMD2E是由4q12的SGCB基因6号外显子突变,导致编码β-肌聚糖功能异常而致病[17],至今共有70余种SGCB致病性突变,也是因为研究报道的总病例数比较少而不能确定致病性的热点突变。例5表现为肢体乏力伴活动耐量下降,肌酸激酶增高伴肌电图异常,SGCB基因的纯合变异(c.1A>G,p.M1?),此位点突变影响蛋白质翻译过程的启动而致病,故确诊LGMD2E。
随着二代测序技术的广泛应用,很多肌营养不良患儿根据临床表现、血清肌酸激酶改变、肌电图,结合基因检查就可确诊并分型,仍不能明确诊断者进行创伤性的肌活检帮助诊断[1,18-19]。本组患儿经临床资料和基因检查均明确诊断,故未进行肌活检。
目前LGMD 的各个类型都缺乏有效的治疗方法,仍以多学科联合的临床康复管理为主。如关节活动锻炼、跟腱挛缩松解、矫形器具及康复锻炼,尽量长时间地维持肢体活动和功能,使生活质量最优化,并预防和管理并发症[5,20]。近年来,多种小分子物质及基因治疗等显示了良好的治疗前景[5,20]。重组人MG53蛋白可有效提高LGMD2B模型小鼠的肌肉膜完整性[21],在疾病进展早期使用FTY 720 可有效的缓解LGMD2C模型小鼠的肌肉萎缩[22]。多种腺相关病毒载体介导的基因治疗方案在不同的LGMD 模型动物研究中,绝大多数表现出很好的获益,有望成为LGMD的有效治疗手段[23-26]。
综上所述,本组5 例患儿为常染色体隐性遗传LGMD,均经基因检测确诊,患儿的基因变异均遗传自表型正常的杂合突变父母,并发现1 个新的SGCG基因突变位点(c.320 C>T)。提示对肌无力患者,甚至部分尚无肌无力表现,但有不明原因肌酸激酶增高者,基因检测有助于早期诊断。由于目前尚缺乏对LGMD的有效治疗方案,故对家系成员进行遗传生育指导很重要,可最大程度减少类似病例的发生。
猜你喜欢
冰雪运动(2021年1期)2021-07-28
种子(2021年3期)2021-04-12
安徽医专学报(2020年3期)2020-12-25
世界科学技术-中医药现代化(2020年2期)2020-07-25
国际放射医学核医学杂志(2020年2期)2020-05-30
中国中医急症(2019年10期)2019-05-21
中国生育健康杂志(2018年6期)2018-11-13
科技视界(2016年27期)2017-03-14
现代检验医学杂志(2016年4期)2016-11-15
中学生理科应试(2016年7期)2016-05-14
