
雷公藤内酯酮与2,6-二甲基-β-环糊精包合物的制备及性能研究
2019-09-19 02:57晋文李飞杨黄宇蓉杨惠文迟绍明朱洪友雷泽赵焱
分析化学 2019年8期
晋文 李飞杨 黄宇蓉 杨惠文 迟绍明 朱洪友 雷泽 赵焱
摘 要:通过饱和水溶液法制备了雷公藤内酯酮(TN)和2,6二甲基β环糊精(DMβCD)的包合物,采用一维核磁共振氢谱(1H NMR)、二维核磁共振氢谱(2D NMR)、傅里叶变换红外光谱(FTIR)、X射线衍射(XRD)、扫描电子显微镜(SEM)和热重分析(TG)等技术对TN/DMβCD包合物进行表征,推测可能的包合模式。Job曲线法表明, 主客体的包合比为1∶1,采用紫外可见光谱滴定法测定得稳定常数为2891 L/mol。形成包合物后,TN的水溶解度由0.0018 mg/mL增至2.7 mg/mL,提高了150倍。体外释放实验表明,在12 h时包合物累积释放率可达70%,而TN累积释放率达70%需120 h。本研究为制备较高水溶性和低毒性的天然药物活性分子提供了新途径。
关键词:雷公藤内酯酮; 2,6二甲基β环糊精; 包合物; 水溶性; 体外释放
1 引 言
雷公藤内酯酮(Triptonide,TN,结构见图1)为从卫矛科雷公藤中分离出的一种环氧二萜内酯化合物,是雷公藤的主要有效成分之一[1]。研究表明,TN除具有抗炎、免疫抑制、抗生育等多种药理活性外[2],还有优异的抗肿瘤活性,如能有效抑制肺癌细胞、鼻咽癌等多种肿瘤细胞生长、促进细胞凋亡[3~5],在白血病的治疗方面也有显著效果[6]。同时,这个活性分子具有较高的安全性,在较高的剂量下对肝脏和肾脏均无毒副作用,也不会降低血液中红细胞和白细胞的数量[5]。TN作为一种天然活性成分,具有较大的应用价值。但是TN较低的水溶性和生物利用度限制了其进一步开发和临床应用[2,4,7]。Qi等[8]合成了一种能靶向释放的2葡萄糖胺修饰的雷公藤内酯衍生物; Tian等[9]使用聚精氨酸修饰TN,提高了其溶解度,并实现透皮递送; Ling等[10]采用叶酸和十八烷基胺合成出可负载TN的囊泡,提高了TN的水溶解度,并实现其靶向和pH响应释放。然而,这些方法存在成本偏高、合成困难、毒副作用较大等问题,限制了它们的应用。因此,进一步研究新型药物载体以提高TN的水溶性和生物利用度具有重要意义。
环糊精( Cyclodextin,CD)具有疏水的空腔和亲水的表面,使其能包合疏水性药物从而改善药物分子的物化性质。因此,CD及其衍生物作为药物载体已成为超分子化学的研究热点之一[11~23]。近期, 本研究组通过多胺修饰βCD的包合作用增溶槲皮素、花旗松素和黄岑素等黄酮类化合物,使其水溶性分别提高了80~508倍,并且包合后花旗松素的抗氧化活性得到提高[24~26]。2,6二甲基β环糊精(DMβCD)作为一种安全性好的药用辅料,由于低价易得、水溶性较好、低毒性因而在药学领域被广泛应用[19]。然而,采用CD及其衍生物改善TN的水溶性研究未见报道。因此,本研究采用饱和水溶液法制备TN和DMβCD的固态包合物,用紫外光谱滴定法研究DMβCD对TN的包合行为,进一步研究包合物的热稳定性、水溶性和体外释放度。本研究为制备较高水溶性和低毒性的天然药物活性分子提供了新途径。
2 实验部分
2.1 仪器与试剂
UV8000S型紫外可见分光光度计(上海元析公司); DRX500型核磁共振仪(德国Bruker公司); Nicolet IS10型紅外光谱仪(美国Thermo科技有限公司); DX2700型X射线衍射仪(丹东浩元公司); Flex1000型扫描电子显微镜(日本日立高新公司); STA449F3型热重分析仪(德国Netzsch 公司)。
TN(纯度>99%,成都曼斯特生物科技有限公司); DMβCD (分析纯,东京化成工业株式会社)。其它试剂均为分析纯,实验用水为二次蒸馏水。
2.2 TN/DMβCD包合物的制备采用饱和水溶液法制备包合物[25]。准确称取0.01 mmol DMβCD 溶于3 mL水中,称取0.03 mmol TN溶于2 mL无水乙醇中。将TN的乙醇溶液加入到上述DMβCD溶液中,密封,45℃下搅拌反应4天。 在45℃干燥,将所得固体溶于少量水中,采用0.45 μm纤维素膜过滤。所得滤液在50℃下真空干燥,得到白色包合物。
2.3 Job曲线测定
用Job曲线法测定TN/DMβCD包合物的包和比。采用磷酸盐缓冲液(PBS,pH=7.20)配制溶液,保持TN和DMβCD的总浓度为4.0×10-5 mol/L。测定TN/DMβCD摩尔比为0~1的溶液在TN最大吸收波长λmax=204 nm处的吸光度。
2.4 紫外可见光谱滴定实验
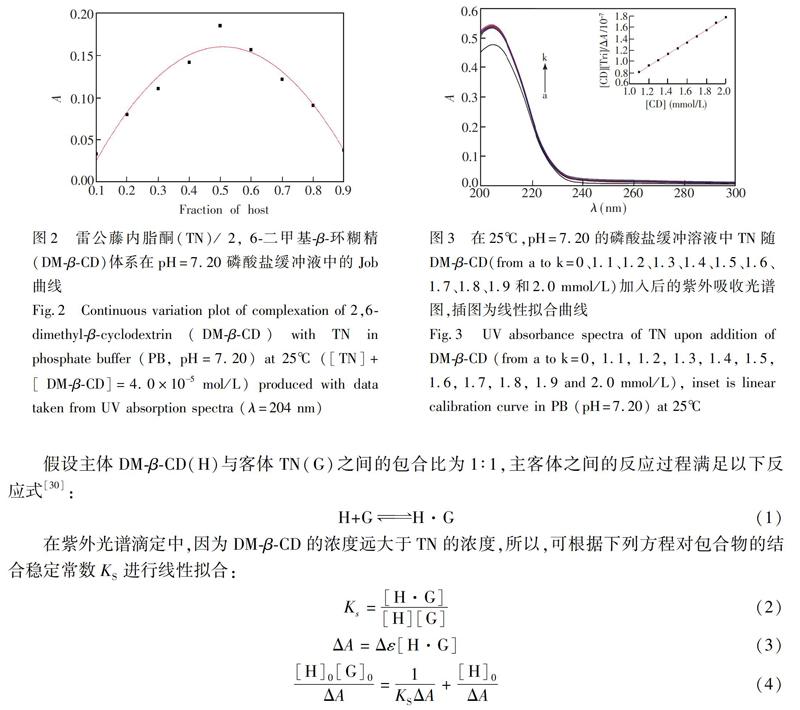
固定TN浓度为5.0×10-5 mol/L,分别加入不同体积的1.0×10-2 mol/L DMβCD溶液,使每份溶液中DMβCD的浓度分别为0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9和2.0 mmol/L。溶剂为乙醇PBS混合液(1∶4, V/V, pH=7.20),测定以上溶液的紫外可见吸收光谱。
2.5 TN的标准曲线
使用无水乙醇配制浓度为0.01、0.02、0.03、0.04、0.05、0.06和0.07×10-3 mmol/L的TN溶液,并测定其在λmax=215 nm处的吸光度, 得其标准曲线方程为A=10200.7143C-0.006199,R2=0.9968 (n=7)。
2.6 TN/DMβCD包合物的水溶性实验
通过饱和水溶液法测定包合物在水中的溶解度[27]。称取过量的包合物溶于400 mL 水中,制备过饱和溶液,在25℃下恒温2 h,期间短暂超声3次,9000 r/min离心10 min,取50 mL上层清液,用无水乙醇定容至5 mL,利用紫外分光光度计测量待测液在215 nm处的吸光度。
2.7 X射线粉末衍射、扫描电子显微镜和热重分析
X射线粉末衍射(XRD)测定条件为(Cu靶Kα为1.54056 ): 工作电压40 kV,电流35 mA,扫描步宽0.02°,采样时间0. 2 s,起止角度为5°~50°。
分别称取10.0 mg TN、10.0 mg DMβCD和10.0 mg TN/DMβCD包合物,用热重分析仪进行测定,设定升温范围20~800℃,升温速率10℃/min,N2流速为70 mL/min。最后分别测得空白样品和包合物的热重分析(TG)曲线。
将适量的TN、DMβCD、TN/DMβCD物理混合物、TN/DMβCD包合物分别固定在载物台上,在电子加速电压为5或3 kV条件下进行扫描电子显微镜(SEM)测试。
2.8 体外释放实验
TN和包合物的体外释放实验在温度为37℃,pH=7.20的PBS溶液中进行[28]。将6 mg TN原料药溶解于10 mL 无水乙醇中,置于洁净的透析袋(截留分子量为1000 Da,美国Spectrumlabs公司)内,然后将透析袋悬浮在含有400 mL PBS溶液的烧杯中, 在搅拌条件下进行透析,转速为100 r/min。 按照预定的时间间隔,从烧杯中取4 mL PBS溶液测定,同时补充相等体积的新鲜PBS溶液到烧杯中。取用载药量相同的包合物溶于水中,采用同样的方法进行测定。
3 结果与讨论
3.1 Job曲线
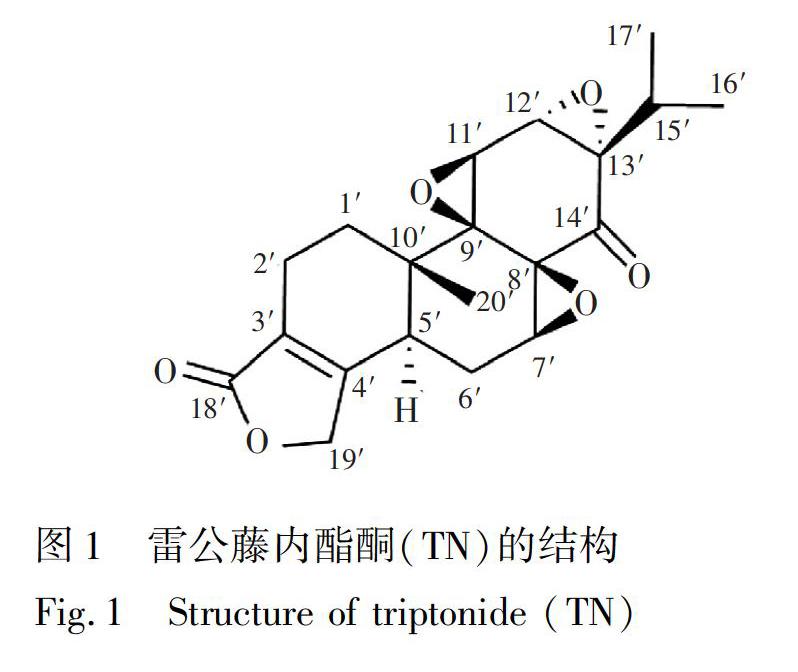
通过Job法测定DMβCD与TN之间的包合比。如图2所示。DMβCD的摩尔分数为0.5时,Job曲线出现最大值,表明DMβCD与TN的包合比为1∶1。
3.2 紫外可见吸收光谱滴定
如图3a所示,TN单独存在时的紫外最大吸收波长204 nm(ε=9526 L/mol·cm),这是由含有α, β不饱和羰基的二萜类化合物ππ*跃迁产生的K带吸收[29]。如图3b~3k所示,随着DMβCD的加入,TN的吸光度值逐渐增大,并伴随最大吸收峰轻微的蓝移,表明DMβCD与TN发生了相互作用,形成了包合物。
3.3 一维核磁共振氢谱(1H NMR)
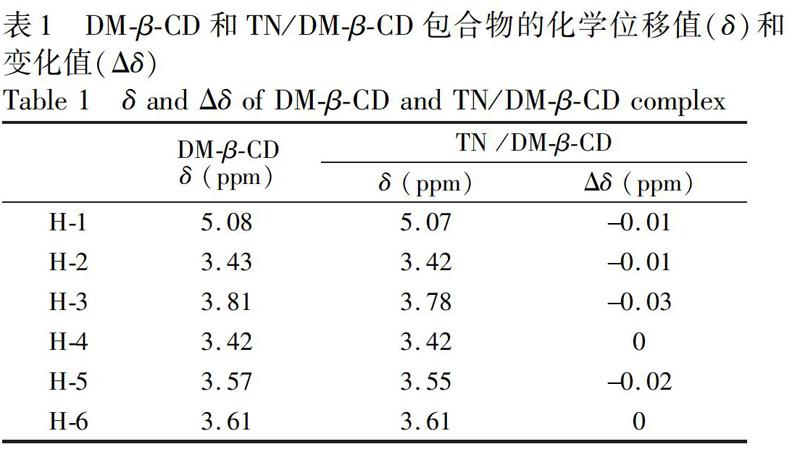
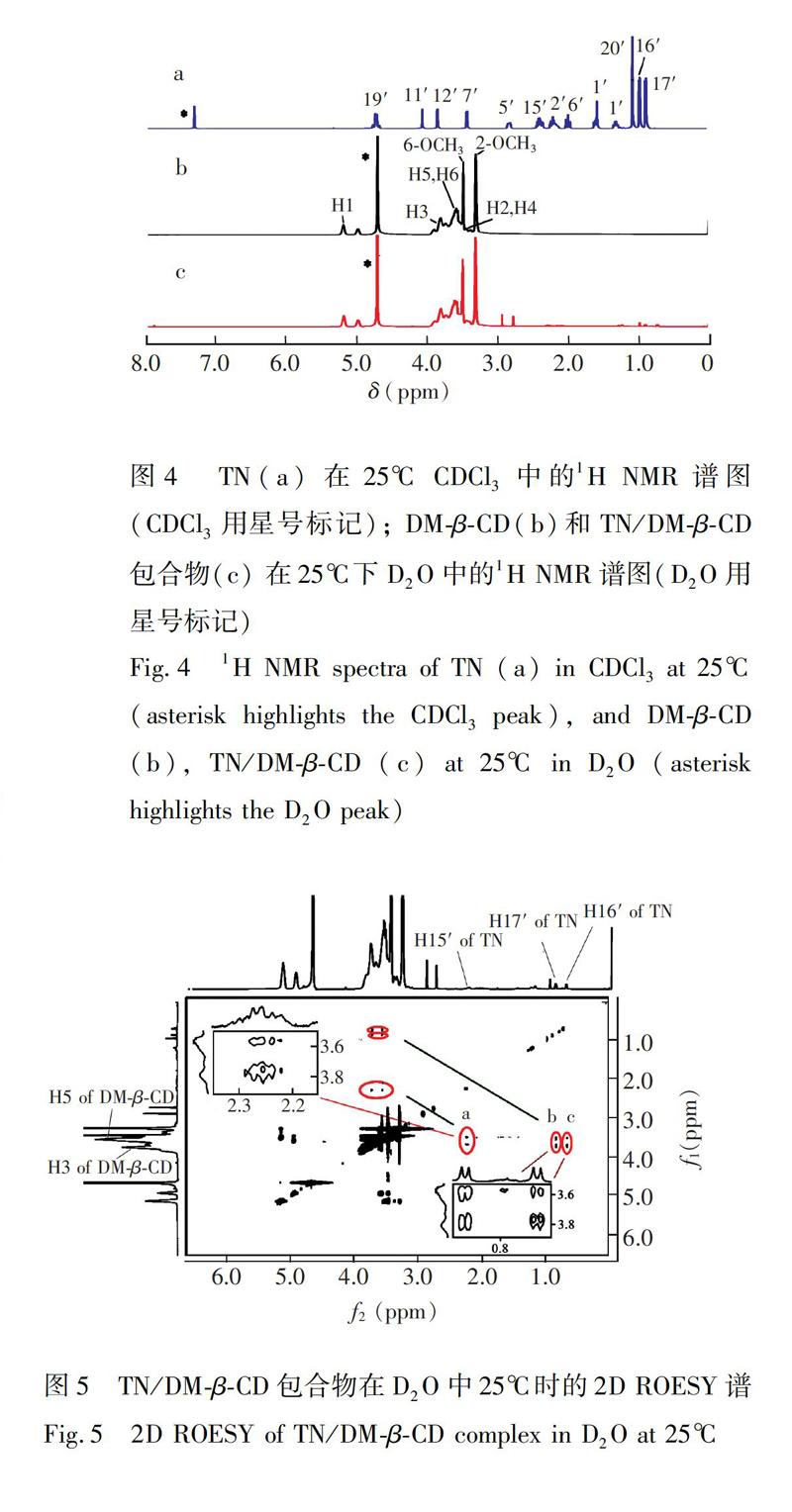
客体分子在与CD包合过程中会导致主客体的化学位移发生变化,这些变化证明了包合物的形成,因此1H NMR技术可作为一种有效的CD包合物的表征方法[14]。如图4所示,a为TN,b为DMβCD,c为TN/DMβCD包合物的1H NMR图。由图4可见,DMβCD的化学位移主要集中在δ=3.0~4.0 ppm处,而TN中特征氢的化学位移主要集中在δ=0.7~3.0 ppm之间,包合物中既包含了DMβCD的特征峰,又包含了TN的特征质子峰。同时,如表1所示,形成包合物后,除H4和H6外,DMβCD中其它质子的化学位移值都向高场轻微移动(Δδ=0.01~0.03 ppm),且H3的Δδ數值大于H5,表明TN分子由H3一侧即大口端进入DMβCD的空腔内。并且,包合物形成后,DMβCD的峰型也发生了微小变化,这表明TN/DMβCD包合物已形成。
3.4 二维核磁共振氢谱 (2D NMR)
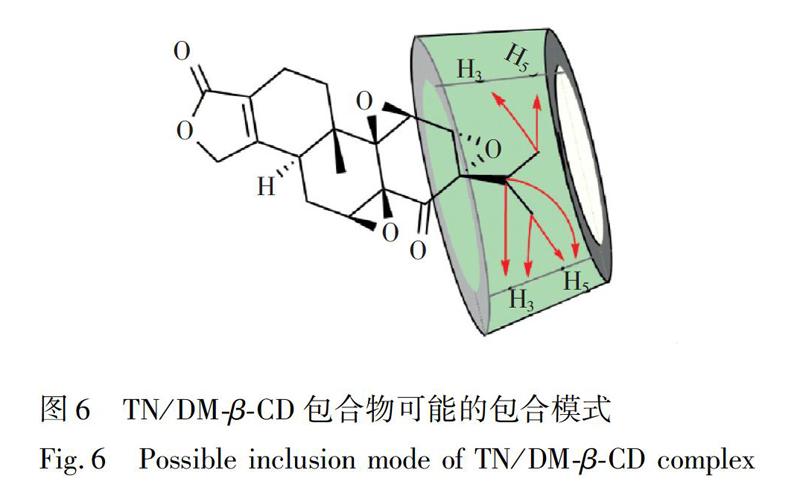
二维旋转坐标系豪泽效应谱(2D ROESY)常用于推测包合物的包合模式。由于CD的H2和H4位于空腔外,H3和H5位于CD的空腔内,而H1容易受到溶剂的影响,故通常用CD的H3和H5与客体质子相互作用的交叉峰进行包合模式分析[19]。图5为TN/DMβCD包合物的2D ROESY谱图,有3个明显的NOE相关点区域:a和c区域显示TN的H16′和H15′与DMβCD的H3、H5均有相关,且与H3的相关性强于H5; b区域显示TN的H17′与DMβCD的H3、H5都有明显的NOE相关点。由于H3位于CD的大口端,H5位于小口端,表明TN的H15′、H16′和H17′质子从大口端进入到CD的空腔中,且H17′进入的更加深入一些,与1H NMR得到的结果一致。结合其包合比为1∶1,分析得到TN/DMβCD包合物可能的包合模式如图6所示。
3.5 红外吸收光谱(FTIR)
使用KBr压片法测定了TN(图7A)、DMβCD(图7B)、TN/DMβCD物理混合物(图7C)和TN/DMβCD包合物(图7D)的红外吸收光谱图。如图7所示,DMβCD和TN的红外光谱图具有明显差异。TN的特征吸收峰主要集中在3471、2984、2939和1764~538 cm1处,而DMβCD的特征吸收峰主要集中在3418、2934、2839和1644~576 cm1。形成包合物后,CD上的OH伸缩振动峰由3418 cm1位移至3414 cm1,且峰型发生了变化,CH伸缩振动峰由2934和2839 cm1位移至2930和2841 cm1,CC伸缩振动由1644 cm1位移至1663 cm1,且峰形发生变化; TN在2984 cm1处的CH伸缩振动峰消失,CO伸缩振动峰由1764和1723 cm1位移至 1759和1727 cm1,1000~400 cm1处的弯曲振动吸收峰消失。与包合物相比,物理混合物的红外光谱图只是二者特征吸收峰的简单叠加,其在3418和1644 cm1处的吸收峰并未产生位移,且峰形无变化,同时在1764 cm1处出现了TN的特征吸收峰。由此推断,形成了TN/DMβCD包合物,而非物理混合物。

3.6 XRD粉末衍射
圖8为TN(图8A)、DMβCD(图8B)、TN/DMβCD物理混合物(图8C) 和TN/DMβCD包合物(图8D)的X射线粉末衍射 (XRD)图谱。TN展现出晶体的形态特征,包合物并未展现晶体的形态特征,而是呈现出与DMβCD更加相似的非晶体形态特征,物理混合物与包合物有明显区别,同时呈现了晶体和非晶体的形态特征,是DMβCD与TN形态特征的简单叠加。与DMβCD相比,包合物的XRD谱图在2θ=12°的衍射峰变得更加尖锐,并在2θ=25°出现了新的衍射峰。形成包合物后,TN在2θ=8°、12°、16°、28°、31°、36°和46° 的衍射峰消失。这些结果表明形成了TN/DMβCD包合物。

3.7 SEM分析
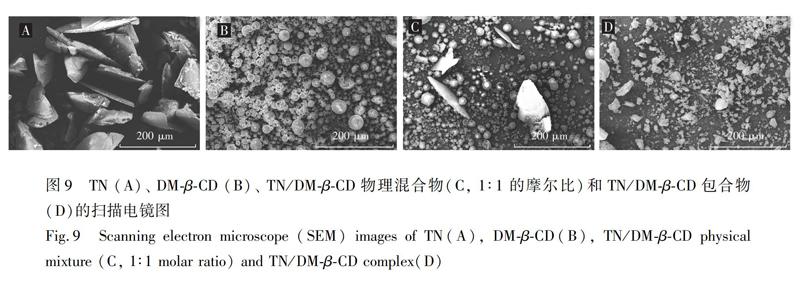
通过SEM观察包合物的外部形貌。如图9所示,TN呈薄片形状(图9A),DMβCD呈小球形状(图9B),物理混合物(图9C)中既有薄片形状的TN,又有小球状的DMβCD,为两者形貌的简单混合,而包合物不同于主客体的形貌,呈现了不规则的块状形貌,并不是简单的主客体形貌的混合(图9D),表明主客体之间形成了包合物。
3.8 热重分析
如图10所示,TN(图10a)有两个失重过程,从19.0℃开始失重,100.8℃失重结束,失重率为7.67%,这一阶段主要是TN失水过程,此后不再失重; 第二阶段是从280.0℃开始失重,585.0℃失重结束,这一阶段是TN 的热分解过程,此过程失重率为64.33%。DMβCD(图10b)有两个失重阶段,第一个阶段是19.5~120.6℃,失重率为8.67%,这一阶段主要是DMβCD失水过程,此后不再失重; 第二阶段是300~420.0℃,失重率为76.50%,这一阶段是DMβCD 的热分解过程。TN/DMβCD包合物(图10c)有两个失重阶段,第一阶段是20.0~100.4℃的失水阶段,失重率为8.56%; 第二个阶段是105.6~298.4℃,这是一个很缓慢的失重过程,这一阶段是受热后,部分TN从βCD空腔缓慢释放的过程,失重率为4.20%,在温度达到300.0℃后,βCDTN包合物开始分解,到405.4℃结束,失重率为68.50%,TN/DMβCD包合物的失重过程不同于主客体,进一步证明包合物的形成。

3.9 包合物水溶性分析
通过饱和水溶液法[27],测得TN在水中的溶解度为0.018 mg/mL,而包合物在水中溶解度达到了2.7 mg/mL。这是由于DMβCD具有疏水的空腔和亲水的表面,使其可通过非共价键相互作用与水溶性差的TN形成包合物,借助DMβCD本身良好的水溶性,使TN的水溶解度提高了150倍。

3.10 体外释放实验
参考文献\[28\]的方法进行TN及其包合物的体外释放实验。TN(a)和TN/DMβCD包合物(b)在PBS溶液(pH=7.20)中的体外释放情况如图11所示,24 h后,包合物累计释放率可达到75%,而TN仅有50%; 12 h后,包合物累积释放率达到70%,而TN累积释放率达70%需要120 h。因此, 形成包合物后,TN的体外释放速率得到显著提高。其原因可能是DMβCD作为一种水溶性药物赋形剂,包合TN后显著提高其水溶解度,有助于难溶性药物的扩散[32]; 另一方面,形成包合物后,TN的结晶度有所降低,也有利于药物的释放[33]。
4 结 论
制备了TN和DMβCD的包合物,研究了DMβCD对TN的包合行为。水溶解度实验表明,形成包合物后,TN的水溶性由0.018 mg/mL提升至2.7 mg/mL, 提高了150倍,在体外释放实验中的12 h累积释放率达到70%,释放的时间缩短了10倍,包合物相比较原料药具有体外释放速度更快的特点。本研究为制备较高水溶性和低毒性的天然药物活性分子提供了新途径。
References
1 Hu D D, Chen X L, Xiao X R, Wang Y K, Liu F, Zhao Q, Li X, Yang X W, Li F. Food Chem. Toxicol., 2018, 115: 98-108
2 Zhang M L, Tan S J, Yu D, Zhao Z, Zhang B, Zhang P, Lv C P, Zhou Q S, Cao Z F. Toxicol. Appl. Pharm., 2019, 365: 1-8
3 Wang S S, Lv Y, Xu X C, Zuo Y, Song Y, Wu G P, Lu P H, Zhang Z Q, Chen M B. Cancer Lett., 2019, 443: 13-24
4 Han H Y, Du L S, Cao Z F, Zhang B, Zhou Q S. Eur. J. Pharmacol., 2018, 818: 593-603
5 Wang Z F, Ma D G, Wang C S, Zhu Z, Yang Y Y, Zeng F F, Yuan J L, Liu X, Gao Y, Chen Y X, Jia Y F. Biomed. Pharmacother., 2017, 96: 757-767
6 Pan Y Y, Meng M, Zheng N N, Cao Z F, Yang P, Xi X D, Zhou Q S. Biochem. Pharmacol., 2017, 126: 34-50
7 He L G, Liang Z Y, Zhao F Q, Peng L F, Chen Z Q. Cell. Mol. Immunol., 2015, 12: 515-518
8 Qi B W, Wang X Y, Zhou Y Y, Han Q, He L, Gong T, Sun X, Fu Y, Zhang Z R. Fitoterapia, 2015, 103: 242-251
9 Tian T, Song Y M, Li K, Sun Y M, Wang Q. Mol. Pharm., 2018, 15: 560-570
10 Ling D, Xia H, Park W, Hackett M J, Song C, Na K, Hui K M, Hyeon T. ACS Nano, 2014, 8(8): 8027-80393
11 Chen X M, Chen Y, Huo X F, Wu X, Gu B H, Liu Y. ACS Appl. Mater. Interfaces, 2018, 10(30): 24987-24992
12 Sun H L, Chen Y, Zhao J, Liu Y. Angew. Chem. Int. Edit., 2015, 54: 9376-9380
13 Guan X R, Chen Y, Wu X, Li P Y, Liu Y. Chem. Commun., 2019, 55: 953-956
14 Li L, Cui G H, Zhao M, Wang Y J, Wang H, Li W, Peng S Q. J. Phys. Chem. B, 2008, 112(38): 12139-12147
15 DENG YingHui, SU LiNa, PANG YanHua, GUO YaFei, WANG Fen, LIAO XiaLi, YANG Bo. Chinese J. Anal. Chem., 2017, 45(5): 648-653
邓颖慧, 苏丽娜, 庞艳华, 郭亚飞, 王 芬, 廖霞俐, 杨 波. 分析化学, 2017, 45(5): 648-653
16 Chen Y, Liu Y. Adv. Mater., 2015, 27: 5403-5409
17 Cheng N, Chen Y, Yu J, Li J J, Liu Y. Bioconjugate Chem., 2018, 29(6): 1829-1833
18 Yang L, Fan S M, Deng G G, Li Y C, Ran X, Zhao H, Li C P. Biosens. Bioelectron., 2015, 68: 617-625
19 Wang L L, Li S S, Tang P X, Yan J, Xu K L, Li H. Carbohyd. Polym., 2015, 129: 9-16
20 SUN Wei, SHE MengYao, MA SiYue, CHEN Jiao, SHI Zhen, LI JianLi. Chinese J. Anal. Chem., 2018, 46(2): 246-253
孙 伟, 厍梦尧, 马思悦, 陈 娇, 史 真, 李剑利. 分析化学, 2018, 46(2): 246-253
21 ZHENG Zhen, CHEN XiuJuan, ZHAO Liang, LI WuHong, HONG ZhanYing, CHAI YiFeng. Chinese Journal of Chromatography, 2017, 35(3): 286-290
鄭 振, 陈秀娟, 赵 亮, 李武宏, 洪战英, 柴逸峰. 色谱, 2017, 35(3): 286-290
22 Han X, Chen Y, Sun H L, Liu Y. Asian J. Org. Chem., 2018, 7: 870-874
23 Zhang Y M, Zhang N Y, Xiao K, Yu Q L, Liu Y. Angew. Chem. Int. Edit., 2018, 57: 8649-8653
24 Zhao L J, Yang S L, Jin W, Yang H W, Li F Y, Chi S M, Zhu H Y, Lei Z, Zhao Y. Aust. J. Chem., 2019, DOI: 10.1071/CH18580
25 Yang S L, Zhao L J, Chi S M, Du J J, Ruan Q, Xiao P L, Zhao Y. J. Mol. Struct., 2019, 1183: 118-125
26 Du J J, Zhao L J, Yang S L, Huang Y R, Chi S M, Ruan Q, Zheng P, Hu R, Zhao Y. J. Incl. Phenom. Macrocycl. Chem., 2019, 93: 203-213
27 Higuchi T, Connors K A. Adv. Anal. Chem. Instrum., 1965, 4: 117
28 Yu S L, Zhang Y J, Wang X, Zhen X, Zhang Z H, Wu W, Jiang X Q. Angew. Chem. Int. Edit., 2013, 125: 1-7
29 GAO JinMing. Phytochemistry. Beijing: Science Press, 2012: 220
高錦明. 植物化学. 北京: 科学出版社, 2012: 220
30 Benesi H A, Hildebrand J H. J. Am. Chem. Soc., 1949, 71(8): 2703-2707
31 Ol'khovicha M V, Sharapovaa A V, Perlovicha G L, Skachilovab S Y, Zheltukhinb N K. J. Mol. Liq., 2017, 237: 185-192
32 Shendea P K, Gaud R S, Bakal R, Patil D. Colloids Surface B, 2015, 130: 105-110
33 Shende P K, Trotta F, Gaud R S, Deshmukh K, Cavalli R, Biasizzo M. J. Incl. Phenom. Macrocycl. Chem., 2012, 74: 447-454
猜你喜欢
保健与生活(2019年22期)2019-11-25
大观(2017年2期)2017-04-07
试题与研究·中考化学(2016年4期)2017-03-28
中学化学(2016年10期)2017-01-07
北方文学·中旬(2016年6期)2016-08-01
科技资讯(2015年19期)2015-10-09
中学生数理化·中考版(2015年11期)2015-09-10
中学化学(2015年5期)2015-07-13
家庭用药(2001年5期)2001-12-24
祝您健康(1992年6期)1992-12-28
