
HPLC-UV-ELSD法同时测定黄芪中黄芪皂苷和黄酮类成分
2019-04-03 08:02史鑫波唐志书刘妍如宋忠兴陈衍斌
天然产物研究与开发 2019年3期
史鑫波,唐志书*,刘妍如,宋忠兴,陈衍斌,苏 瑞,许 刚,王 升
1陕西中医药大学 陕西省中药资源产业化协同创新中心;2陕西省创新药物研究中心,咸阳 712083;3陕西步长制药有限公司,西安 710075;4中国中医科学院中药资源中心,北京 100700
黄芪为豆科植物蒙古黄芪Astragalusmembranaceus(Fisch.)Bge.var.mongholicus(Bge.)Hsiao 或膜荚黄芪Astragalusmembranaceus(Fisch.)Bge.的干燥根[1]。黄芪中含有皂苷类、黄酮类、多糖类、微量元素等多种成分[2-6]。现代药理研究表明,黄芪提取物及其制剂具有调节免疫系统、保护心血管系统、保护神经系统、保护肝肾和抗肿瘤等药理活性[6,7]。黄芪皂苷对心血管和心脏具有保护作用[8-10],黄芪多糖具有抗应激、抗氧化、增强免疫功能等的免疫调节效应[11]。黄芪水溶性黄酮类成分对细胞免疫功能具有促进作用[12,13]。在2015版药典中以HPLC-ELSD和HPLC-UV分别检测黄芪甲苷和毛蕊异黄酮葡萄糖苷的含量,以评价其质量的优劣,但药典中黄芪甲苷含量检测的样品制样方法尤为繁琐。本研究选择黄芪中黄芪皂苷和黄酮类成分作为研究对象,使用大孔树脂制备的供试品溶液,黄芪皂苷和黄酮类成分较高,且制备方法简便,重复性好。本实验所建立的HPLC-UV-ELSD 方法简便、准确,可同时测定黄芪7种有效成分含量,为有效控制黄芪的内在质量提供参考和依据。
1 仪器与材料
1.1 仪器
Waters e2695高效液相色谱仪,配备2489UV/Vis检测器和2424 EIS检测器(沃特世科技有限公司);空气压缩机(GA-10B北京中兴正科科技发展有限公司);KQ-300DE型数控超声波清洗器(昆山市超声仪器有限公司);十万分之一电子天平(赛多利斯科学仪器有限公司)。
1.2 材料
黄芪甲苷(批号为20150517,质量分数≥98.0%);黄芪皂苷Ⅰ(批号为20160601,质量分数≥98.00%);黄芪皂苷Ⅱ(批号为20160327,质量分数≥98.0%);黄芪皂苷Ⅲ(批号为20160503,质量分数≥98.00%)均购于宝鸡市辰光生物科技有限公司,毛蕊异黄酮葡萄糖苷(批号为111920-201505,质量分数≥97.10%)购于中国食品药品检定研究院,毛蕊异黄酮(批号为16120911,质量分数≥99.84%);刺芒柄花苷(批号为17041101,质量分数≥98.50%)均购于中国科学院成都生物研究所。
实验用黄芪饮片购自陕西兴盛德药业有限责任公司,经陕西中医药大学陕西省中药资源产业化协同创新中心刘世军高级工程师鉴定为蒙古黄芪Astragalusmembranaceus(Fisch.)Bge.var.的干燥根。实验用乙腈和甲酸均为色谱纯;水为娃哈哈纯净水;其余试剂为分析纯。
2 方法与结果
2.1 色谱条件
色谱柱为Agilent 5 TC-C18(250 mm×4.6 mm,5 μm);流动相为乙腈-0.3%甲酸水,梯度洗脱,洗脱条件为:0~10 min,15%~40%乙腈;10~20 min,40%乙腈;20~30 min,60%~85%乙腈;30~40 min,85%~15%乙腈;流速1.0 mL/min;进样量10 μL;检测方式:UV/ELSD串联监测;UV:检测波长254 nm;柱温30 °C;ELSD参数:增益100,漂移管温度70 °C,气压0.7 Mpa(见图1)。
2.2 对照品溶液制备
分别称取黄芪甲苷、黄芪皂苷Ⅰ、黄芪皂苷Ⅱ、黄芪皂苷Ⅲ、毛蕊异黄酮葡萄糖苷、刺芒柄花苷、毛蕊异黄酮适量,精密称定,加甲醇溶解制成各成分质量浓度分别为1.78、0.55 、0.40、1.14、0.40、0.20、0.15 mg/mL的混合标准品工作储备溶液。
2.3 供试品溶液制备
2.3.1 大孔树脂制备供试品溶液
黄芪饮片,在60 °C下干燥3 h,粉碎(过4 号筛),称取黄芪粉末约4 g,置索氏提取器中,加入80.0 mL甲醇,冷浸过夜,加热回流4小时,提取液回收并浓缩至干,将浓缩提取物加水5 mL,微热溶解,通过D 101大孔树脂脂柱(内径1.5 cm、长12 cm),以水50 mL洗脱,弃去水液,再用20%乙醇50 mL 洗脱,弃去洗脱液,继续用85% 乙醇100 mL 洗脱,收集85%乙醇洗脱液,浓缩至干,用甲醇溶解并转移至5 mL量瓶中,加甲醇至刻度,摇匀,过0.22 μm微孔滤膜,即得供试品溶液1[14]。
2.3.2 固相萃取制备供试品溶液
取2.3.1项下浓缩提取物加水5 mL,溶解,过固相萃取柱(500 mg/6 mL,先用20 mL 甲醇和5 mL 水预处理),以10 mL 水洗涤,弃去水洗液,后用20%乙醇溶液10 mL 洗涤,弃去洗涤液。用85%乙醇40 mL 洗脱,收集洗脱液,浓缩至干,加甲醇使其溶解并定容至5 mL 量瓶中,滤过,取续滤液,过0.22 μm微孔滤膜,即得供试品溶液2[15]。
2.3.3 经过碱化(KOH)处理的供试品溶液
称取黄芪粉末约4 g,置索氏提取器中,加2%的 KOH 80.0 mL 甲醇,加热回流4小时,提取液回收溶剂并浓缩至干,将浓缩提取物加水5 mL,溶解,通过D101 大孔树脂柱(内径1.5 cm、长12 cm),以水50 mL 洗脱,弃去水液,再用20%乙醇50 mL 洗脱,弃去洗脱液,继续用85% 乙醇100 mL 洗脱,收集85%乙醇洗脱液,蒸干,用甲醇溶解并转移至5 mL量瓶中,加甲醇至刻度,过0.22 μm微孔滤膜,即得供试品溶液3[16]。
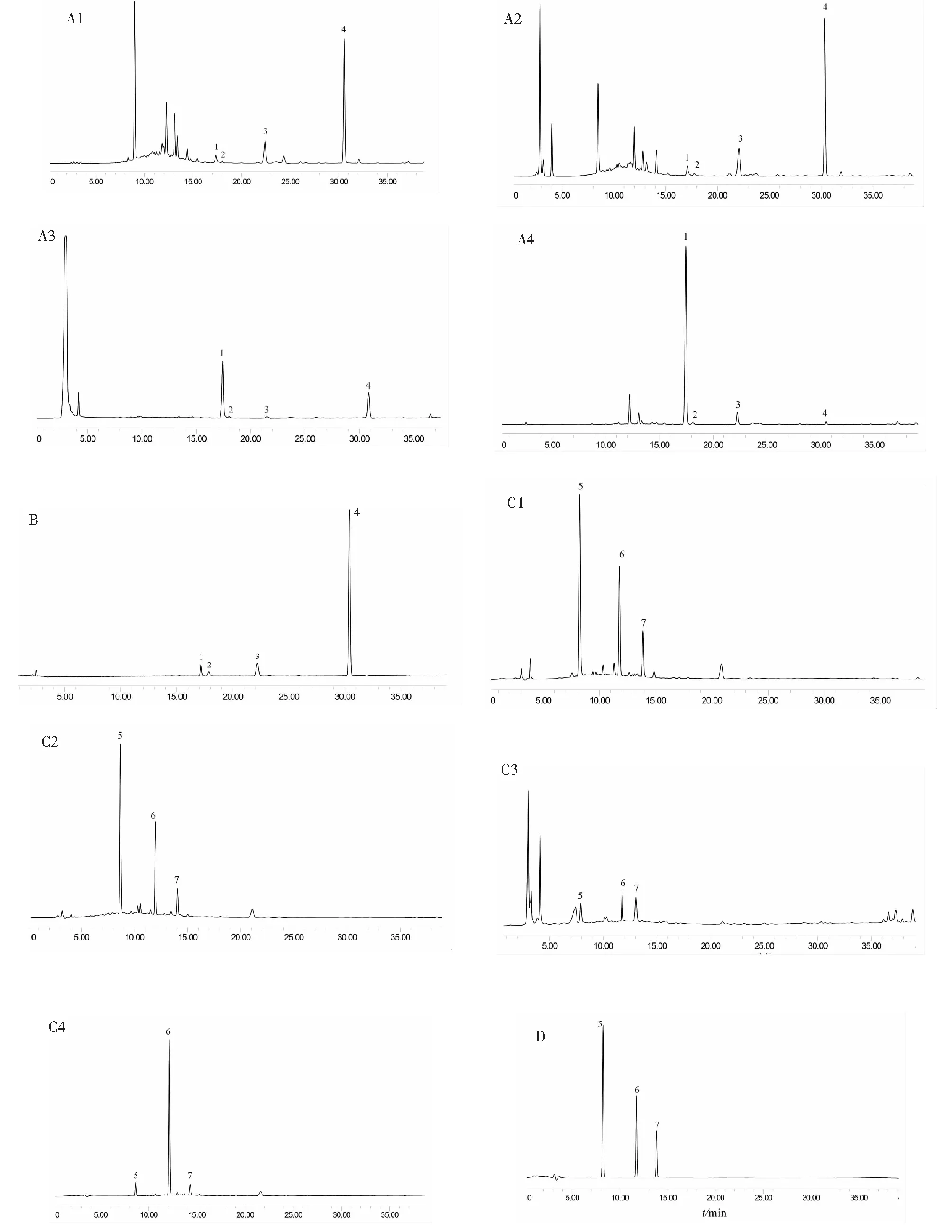
图1 黄芪供试品溶液1-4(A1-A4)和混合黄芪皂苷对照品(B)的HPLC-ELSD色谱图Fig.1 HPLC-ELSD chromatograms of Astragali Radix sample 1-4(A1-A4) andsaponins mixed reference substances (B)注:1.黄芪甲苷;2.黄芪皂苷Ⅲ;3.黄芪皂苷Ⅱ;4.黄芪皂苷Ⅰ。Note:1.astragaloside I;2.astragaloside II;3.astragaloside III;4.astragaloside IV.
2.3.4 经过碱化(氨水)处理后萃取的供试品溶液
取2.3.1项下浓缩提取物加水5 mL,溶解,用水饱和的正丁醇振摇提取4次,每次40 mL,合并正丁醇液,用氨试液充分洗涤2次,每次40 mL,弃去氨液,将正丁醇液蒸干,提取液回收溶剂并浓缩至干,将浓缩提取物加水5 mL,微热溶解,放至室温,通过D 101型大孔吸附树脂柱,以水50 mL洗脱,弃去水液,再用40 %乙醇30 mL洗脱,弃去,再用70%乙醇80 mL洗脱,收集洗脱液,蒸干,加甲醇溶解,转移至5 mL量瓶中,加甲醇至刻度,过0.22 μm微孔滤膜,即得供试品溶液4[1]。
2.4 方法学考察
2.4.1 线性关系的考察
按2.1项下设定色谱条件,分别精密吸取2.2项下混合标准工作溶液1、5、10、15、20、25、30 μL进样,进行色谱分析,毛蕊异黄酮葡萄糖苷、 刺芒柄花苷、毛蕊异黄酮以色谱峰峰面积(Y)为纵坐标,以进样量为横坐标(μg,X),进行线性回归,绘制标准曲线,黄芪甲苷、黄芪皂苷Ⅲ、黄芪皂苷Ⅱ、黄芪皂苷Ⅰ以峰面积积分值(Y)的对数值为纵坐标,分别以对照品进样量(μg,X) 的对数值为横坐标,绘制标准曲线。7种成分的线性方程、相关系数、线性范围见表1。

表1 黄芪中7种成分的线性方程、线性范围、相关系数
2.4.2 精密度试验
取供试品溶液1,按2.1项下设定色谱条件连续进样6次,每次进样10 μL,结果黄芪甲苷、黄芪皂苷Ⅲ、黄芪皂苷Ⅱ、黄芪皂苷Ⅰ、毛蕊异黄酮葡萄糖苷、刺芒柄花苷、毛蕊异黄酮的峰面积RSD峰面积为1.37%、2.76%、1.07%、0.64%、0.54%、0.81%、2.23%,表明仪器精密度好。
2.4.3 稳定性考察
取供试品溶液1,按2.1项下设定色谱条件进行测定,记录各色谱峰峰面积,分别在配制后0、2、4、8、12、24 h,各进样10 μL,结果黄芪甲苷、黄芪皂苷Ⅲ、黄芪皂苷Ⅱ、黄芪皂苷Ⅰ、毛蕊异黄酮葡萄糖苷、刺芒柄花苷、毛蕊异黄酮的峰面积RSD峰面积为0.98%、1.53%、1.30%、0.77%、5.01%、0.49%、2.21%,表明供试品溶液在24小时内稳定性良好。
2.4.4 重复性试验
取同一批黄芪粉末6份,按2.3.1项下方法制备供试品溶液,按2.1项下设定色谱条件进行测定,结果黄芪甲苷、黄芪皂苷Ⅲ、黄芪皂苷Ⅱ、黄芪皂苷Ⅰ、毛蕊异黄酮葡萄糖苷、刺芒柄花苷、毛蕊异黄酮的峰面积RSD峰面积为3.63%、4.82%、1.87%、2.52%、3.68%、4.81%、1.56%,表明方法重复性良好。
2.4.5 加样回收率试验
分别精密称取已知有效成分含量的药材2.0 g,分别加入各对照品适量,按2.3.1项下方法制备供试品溶液6份。按2.1项下设定色谱条件进行测定,计算加样回收率,结果见表 2 所示。

表2 加样回收率实验结果(n=6)
2.5 样品含量测定
按 2.3项下方法制备四种不同的供试品溶液,按2.1项下设定色谱条件进行测定,含量测定结果见表3。

表3 4种黄芪供试品中7种黄酮和皂苷成分含量测定结果(mg/g, n=3)
四种供试品种中,皂苷总量从高到低:供试品1>供试品2>供试品3>供试品4。黄酮总量从高到低:供试品1>供试品2>供试品4>供试品3。
2.6 主成分分析
主成分分析(Principal component analysis)是一种有效的降低数据维数的方法,适合于高维光谱数据的定量及定性分析[17]。为综合评价不同处理方法对黄芪有效成分的影响,利用综合评价法以确定适宜的处理方法[18],通过SPSS 19.0统计软件对对已测定的黄芪皂苷和黄酮类成分进行了主成分分析。得到主成分总变量和主成分载荷矩阵(表4、5),前2个主成分的特征值均大于1,计算得到前2个主成分(PC1和PC2)累积方差贡献值(96.79%),能够较客观地反映不同处理方法黄芪样品的内在质量。PC1主要反映了黄芪甲苷、黄芪皂苷Ⅰ、毛蕊异黄酮葡萄糖苷、毛蕊异黄酮的信息,PC2反映了黄芪皂苷Ⅲ、黄芪皂苷Ⅱ和刺芒柄花苷的信息。
采用2个主成分对黄芪不同处理方法进行评价。以各主成分因子得分与方差贡献率乘积之和相加,可反映各黄芪供试品各类成分总因子得分值F,其综合评价函数为F=0.815 14×F1+0.152 44×F2。按综合评价函数计算出不同样品的综合得分F(见表6)。由综合得分可知供试品1得分最高,供试品2次之,与黄酮和皂苷成分含量测定结果一致。
3 讨论
本研究采用4种不同的处理方法对黄芪的有效成分进行提取,采用HPLC-UV-ELSD同时测定其中7种成分含量,更能简便、直观反映黄芪药材的质量,可为黄芪的多组分同时检测及质量标准改善提供新思路和参考数据。
由供试品3和供试品4的结果表明,碱化处理的顺序会对黄芪皂苷是有一定影响。首先对药材进行碱化处理,再进行分离提取,可使黄芪皂苷之间的相互转化更充分,更彻底。结合黄芪皂苷的结构可得,黄芪皂苷Ⅰ具有2个乙酰基,黄芪皂苷Ⅱ有1个乙酰基,在遇到碱时黄芪皂苷Ⅰ容易脱落乙酰基而转化成黄芪皂苷Ⅳ(或其它类黄芪皂苷)。黄芪皂苷Ⅲ因不具有乙酰基,所以处理方法对其影响较小。黄芪皂苷Ⅰ、黄芪皂苷Ⅱ转化为黄芪甲苷,但这种转化受碱性条件和提取条件等影响,难以实现完全转化,不能客观真实地反映黄芪皂苷的实际含量[19]。综合含量测定结果和PCA分析结果可以得出,未经碱化处理的黄芪中黄芪皂苷和黄酮类成分含量更高,大孔树脂可提高皂苷和黄酮类成分的富集。从四种供试品的UV-ELSD分析谱图可以得出,经处理和提取后,部分相关色谱峰变化比较明显,即黄芪的成分发生了较大的变化,就此类成分的分析将在下一步的实验中进行研究。

表4 主成分的特征值及贡献率

表5 旋转变换后的因子载荷矩阵
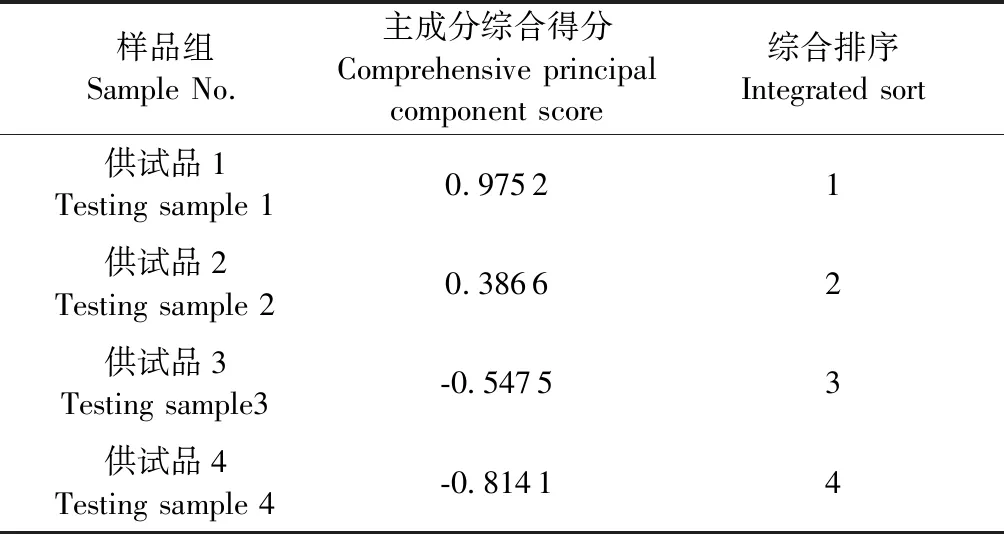
表6 综合主成分评价结果
猜你喜欢
世界最新医学信息文摘(2021年82期)2021-07-13
世界科学技术-中医药现代化(2021年12期)2021-04-19
山东医药(2021年3期)2021-02-24
中国现代医学杂志(2020年11期)2020-09-25
安徽中医药大学学报(2020年2期)2020-04-21
中国老年学杂志(2019年20期)2019-10-17
中国外汇(2019年10期)2019-08-27
中国外汇(2019年10期)2019-08-27
中国外汇(2019年22期)2019-05-21
中国外汇(2019年21期)2019-05-21
