
SNP芯片联合质谱筛检胃癌相关基因3´UTR区SNPs的病例-对照研究
2018-10-16 01:46韩仁杰吴传城刘宝英郭赛熊陈
癌变·畸变·突变 2018年5期
韩仁杰吴传城刘宝英 *郭赛熊陈 昱
(1. 福建医科大学公共卫生学院,福建福州 350108;2. 福建省莆田市仙游县医院,福建 莆田 351200)
胃癌是世界范围内最常见的恶性肿瘤,居消化系统肿瘤的首位[1]。加强对胃癌高发区高危人群发生胃癌危险性的监控,对胃癌的防治有重要的现实意义[2]。近年来大量的流行病学研究发现某些基因3´非翻译区(untranslated region,UTR)有许多单核苷酸多态性位点(single nucleotide polymorphisms,SNPs)与肿瘤的易感性显著相关,研究表明基因3´UTR的SNPs与人类疾病相关的机制之一,是其存在miRNA的结合位点[3],这些SNPs的存在有可能影响miRNA-mRNA配对的有效性以及结合数量,从而通过影响基因的转录后调控,参与肿瘤的发生发展[4]。本研究采用SNP芯片联合飞行时间质谱技术筛检福建省仙游县胃癌患者血液中差异的消化道肿瘤相关基因3´UTR区SNPs,采用病例-对照研究分析芯片筛选出来的SNPs与胃癌易感性的关系。
1 材料与方法
1.1 研究方法与对象
采用1∶1配对的病例-对照研究,胃癌病例来源于福建省仙游县医院的胃癌新发病例。大样本纳入标准为:经手术或内窥镜取得组织标本,经病理确诊的新发病例;确诊日期为2013年4月至2017年10月;在仙游本地居住10年以上。芯片组纳入标准是在此基础上选择男性,年龄50~70岁之间,经病理确诊为胃腺癌的患者。排除标准:经病理确诊为胃部炎症、良性病变及病情危重或不能清晰回答问题者及肿瘤继发病例和复发病例。
健康对照纳入标准:按性别、年龄、地区与病例进行配对,其中芯片健康对照选择年龄±3岁与病例进行配对,大样本健康对照选择年龄±5岁与病例进行配对。选择在仙游本地居住10年以上者。排除标准为:胃癌或慢性萎缩性胃炎的直系家属。
以上标本均要求资料和血样完整,最后SNP芯片筛选阶段我们纳入96例胃癌患者和96例对照,均为男性,年龄分布在52~71岁之间。其中病例组平均年龄(63.84±4.64)岁,对照组平均年龄(63.84±4.66)岁。
在验证阶段纳入确诊胃癌病例622例,正常对照622例,其中男性466对,女性156对。病例组由312例贲门癌患者和310例非贲门癌患者组成。病例组年龄分布在39~89岁之间,平均年龄为(67.32±9.72)岁;对照组年龄分布在41~87岁之间,平均年龄为(67.22±9.65)岁。经均衡性检验,病例组与对照组在年龄、性别、职业等方面差异均无统计学意义(P>0.05)。病例组中高中及以上的文化程度比例低于对照组,已婚的比例高于对照组,差异有统计学意义(P<0.05)。
1.2 资料收集
采用统一设计的调查表,进行面对面的访谈式调查。病例的病史资料经患者本人及医院同意后通过摘录电子病历获得。调查内容包括研究对象的年龄、性别、文化程度、婚姻状况、职业史等相关因素,同时采集病例及对照人群的空腹外周静脉血5 mL,抽取的血样置于EDTA抗凝管,3 000 r/min离心10 min后,分装成血浆、白细胞、红细胞,置-80 ℃低温冰箱保存。本研究涉及的调查数据及人体数据,均已通过福建医科大学生物医学研究伦理委员会的审查,符合医学伦理相关规定和要求。
1.3 芯片基因分型原理与方法
1.3.1 基因分型原理 本过程通过Affymetrix Axiom SNP芯片对芯片中消化道肿瘤相关基因3´UTR区SNPs进行基因分型,基因芯片分型技术采用酶介导及单碱基测序的全自动化流程。
1.3.2 消化道肿瘤相关基因3´UTR区SNPs的筛选方法 利用KEGG-PATHWAY数据库(http://www.kegg.jp/kegg/pathway)搜索食管癌、胃癌、肠癌、肝癌等消化道肿瘤相关基因。根据dbSNP数据库(https://www.ncbi.nlm.nih.gov/snp/)筛选位于这些基因3´ UTR的SNPs。将筛选出的SNPs与芯片位点做交集。再利用SPSS和Excel软件对所有的消化道肿瘤相关基因3´UTR区SNPs进行卡方检验,选取 P<0.10的位点;根据千人基因网站(https://www.ncbi.nlm.nih.gov/variation/tools/1000 genomes/)查找各位点的最小等位基因频率(minor allele frequency,MAF),选择MAF在0.15~0.40的位点;对各多态性位点进行Hardy-Weinberg遗传平衡度检验,选取基因型频率分布在健康对照人群中符合Hardy-Weinberg遗传平衡定律的位点进行后续实验分析。
1.3.3 大样本人群中miRNA基因分型原理与方法 将芯片中筛选得到的5个SNPs位点区域的DNA模板通过PCR技术扩增,再使用特异的延伸引物与PCR产物进行单碱基延伸反应。通过基质辅助激光解吸附电离飞行时间质谱分析质谱技术(MALDI-TOF-MS),检测延伸产物相对分子质量,应用专用的分析软件,通过判断分子大小的差异而进行SNP分型检测。
1.4 PCR扩增引物设计
对芯片选取的5个SNP位点进行基因型鉴定,使用Sequenom公司Genotyping Tools及Mass ARRAY Assay Design软件设计待测SNP位点的PCR扩增引物及单碱基延伸引物,见表1。
1.5 质量控制

?
按以下标准进行质量控制:①所有病例均经过病理确诊;②芯片分型过程中使用评估信号值分布与背景信号值的差异(Dish QC)对样品进行质控,Dish QC低于0.82的样品不纳入后续的分型中;③基因型的判读采用盲法;④芯片检测与MALDI-TOF-MS检测进行结果一致性比较;⑤少数样本由于DNA质量或者浓度问题,未能检测出基因型的样本予以剔除。
1.6 统计分析
所有实验室基因型数据经过核实后建立数据库,采用 χ2检验对病例组和对照组的一般人口学资料进行均衡性检验;采用 χ2检验计算对照组的各基因多态性位点分布是否符合Hardy-Weinberg平衡定律(H-W平衡);采用Cox Regression命令拟合条件Logistic模型对单个位点的基因型、等位基因进行单因素Logistic分析,计算其比值比(odds ratio, O R)及95%可信区间(confidential interval, C I)。以上分析均采用SPSS 21.0软件包完成。所有 P值基于双侧检验,统计学检验水准α=0.05。
2 结 果
2.1 芯片中消化道肿瘤相关基因3´UTR区SNPs筛选情况
按照消化道肿瘤相关基因3´UTR区SNPs筛选策略,最终选出5个与胃癌相关的消化道肿瘤相关基因3´UTR区SNPs,见图1。分别为表皮生长因子受体(epidermal growth factor receptor, EGFR) EG FR rs884225 C>T, EGFR r s10277413 G>T,F A S r s1468063 C>T,MSH2 rs17502941 A >G,M SH2 r s11125144 A >G,见表2。
2.2 消化道肿瘤相关基因多态性与胃癌关联性分析
本次选取的5个SNPs位点的基因型频率分布在对照组中符合H-W 平衡(P >0.05)。经婚姻状况、文化程度等危险因素校正的条件Logistic回归分析未发现两位点与胃癌易感性相关,见表3。对病例-对照的分层分析显示rs884225中含有T等位基因的基因型(CT+TT)与贲门癌相关(O R= 0.67,95% C I:0.46~0.99),而其他位点与贲门癌、非贲门癌的风险关联无统计学意义(P>0.05),见表4。
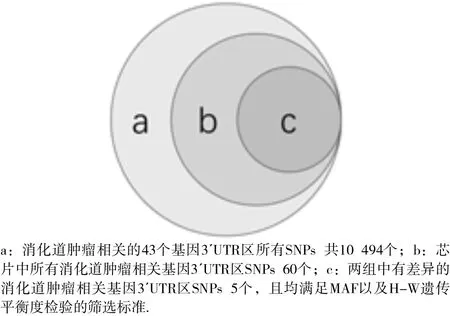
图1 消化道肿瘤相关基因 3 ´UTR区 S NPs筛选情况图
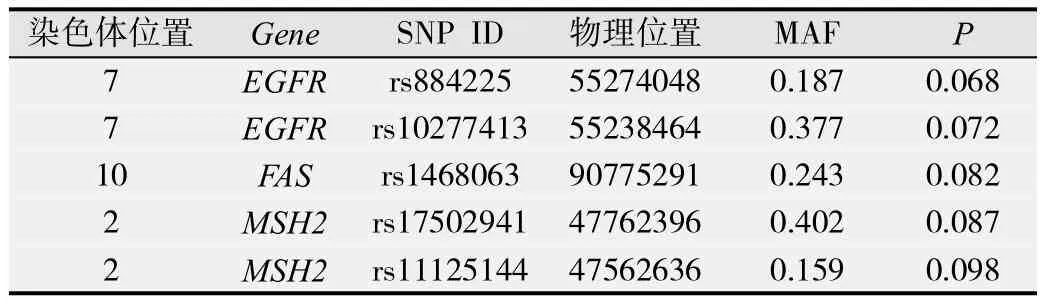
表2 有差异的消化道肿瘤相关基因3 ´UTR 区 S NPs 及其基本信息
2.3 单体型分析
2.3.1 EGFR基因单体型分析 我们所研究的EGFR基因2个多态性位点共可产生4个单体型SNP位点,排列顺序依次为rs884225、rs10277413,结果单体型1~4在病例和对照组的分布差异均无统计学意义(P>0.05)。见表5和表6。
2.3.2 MSH2基因单体型分析MSH2基因2个多态性位点共可产生4个单体型SNP位点,排列顺序依次为rs17502941、rs11125144,结果单体型1~4在病例和对照组的分布差异均无统计学意义(P>0.05)。见表7和表8。
3 讨 论
近年来发现miRNA在转录后水平调控基因的表达进而参与调控肿瘤的发生发展。而miRNA与SNP均在分子水平上对肿瘤的发生发展产生一定的调控作用[5],因此探索miRNA与SNP之间的相互作用对肿瘤的作用不容忽视。miRNA编码基因本身可存在SNPs位点[6],而在靶基因mRNA 3´UTRs的miRNA结合位点上也有可能存在SNPs位点[7],单个碱基的改变均有可能影响miRNA与mRNA配对的有效性以及结合数量,从而在转录后水平改变靶基因的蛋白质表达[8]。由于miRNA片段小,发生在miRNA编码基因上的SNP数量少,而miRNA与靶基因结合位点上的SNPs可减弱相应miRNA对靶基因的翻译抑制效应,增强靶基因表达或者形成新的miRNA结合位点,从而抑制靶基因的表达[9]。若该基因与胃癌的发生发展密切相关,则发生在其3´非翻译区的SNP可影响胃癌的易感性。目前对于miRNA靶位点的SNP与肿瘤的研究多采用生物信息学预测的方法[10],由于预测结果缺乏一定的可靠性,因此在本研究中我们采用SNP芯片对消化道肿瘤相关基因3´UTR区SNPs位点进行关联分析,然后通过飞行时间质谱法验证,最后发现1个SNP位点与贲门癌易感性相关。

表3 消化道肿瘤相关基因3´UTR区SNPs与胃癌的关联程度
本次研究首先通过生物信息学方法筛选出43个消化道肿瘤相关基因共计10 494个3´UTR区SNPs位点。但其中包含了许多SNPs位置相同但名称不一致的位点,我们为了不遗漏地筛选出消化道肿瘤相关基因3´UTR区SNPs,将所有通过生物信息学筛选出的SNPs位点与SNP芯片中的位点做交集,发现有9个消化道肿瘤相关基因3´UTR区SNPs位点在芯片中。通过卡方检验分析后发现有5个SNPs位点基因分型在病例对照组中分布差异有统计学意义,然后对该5个SNPs位点进行大样本验证,最后筛查出来的5个SNPs中发现EGFR rs884225与贲门癌的易感性相关(P<0.05),而其他位点与胃癌发病风险无关联(P>0.05)。有文献报道EGFR rs884225[11]及 F AS rs1468063[12]均与胃癌的发病风险密切相关,而其他位点还未见相关文献报道。
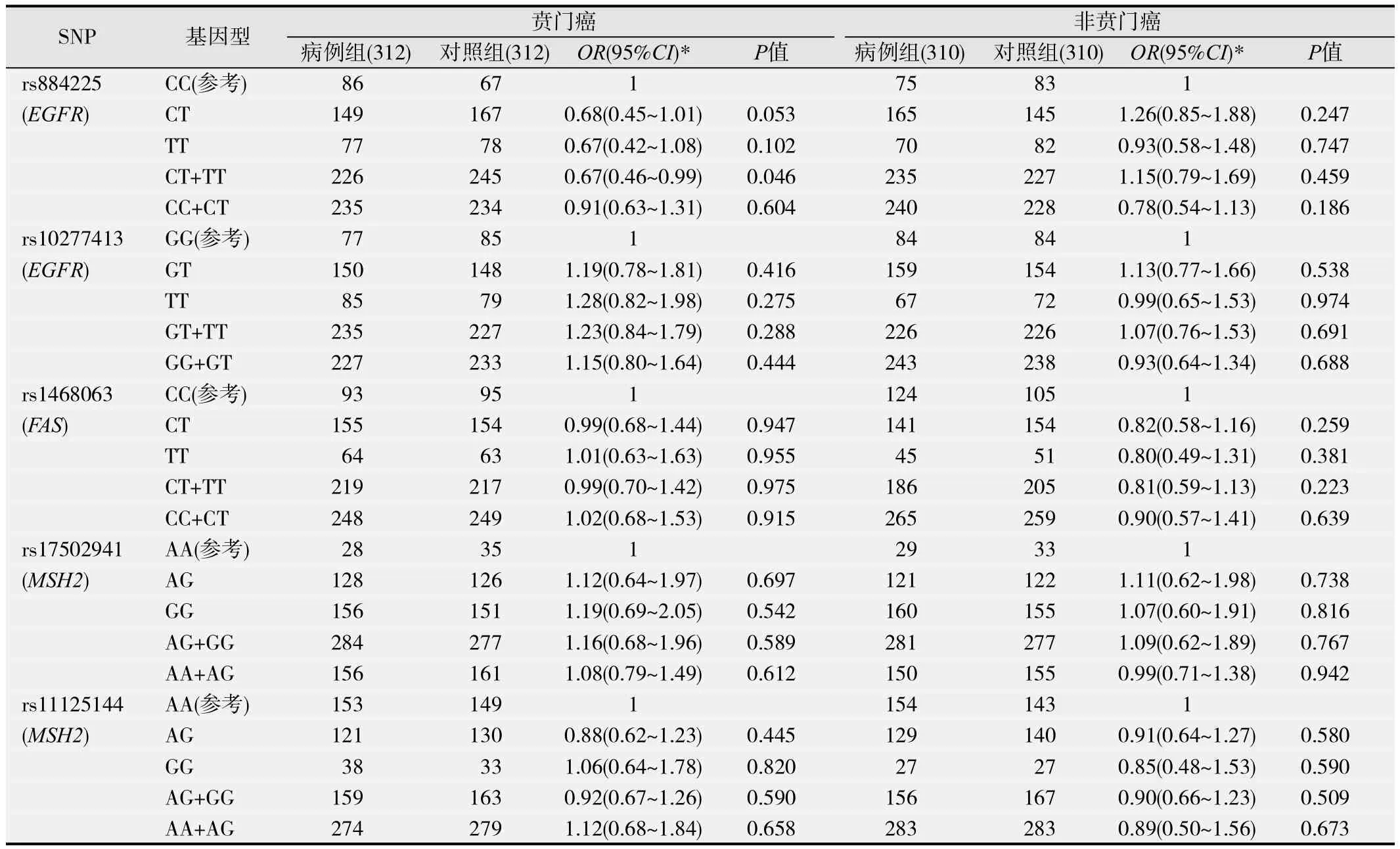
表4 消化道肿瘤相关基因3´UTR区SNPs与不同胃癌部位的关联程度

表5 EGFR不同单体型在胃癌人群中的分布情况

表6 EGFR不同单体型在贲门癌、非贲门癌人群中的分布情况
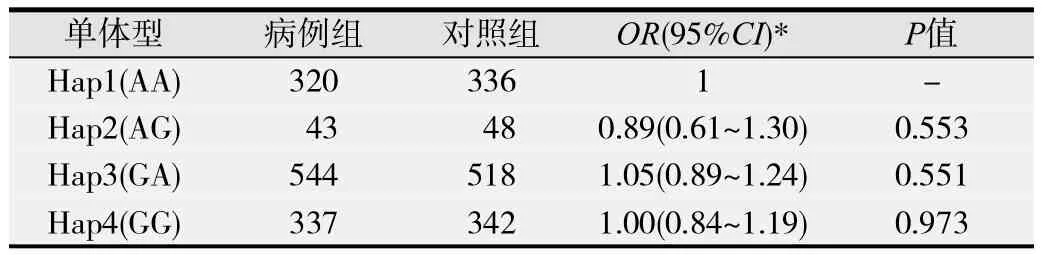
表7 MSH2不同单体型在胃癌人群中的分布情况
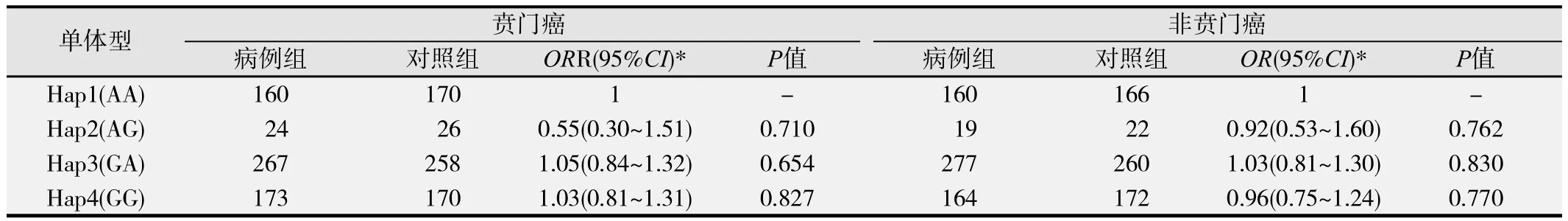
表8 MSH2不同单体型在贲门癌、非贲门癌人群中的分布情况
表皮生长因子受体(EGFR)是HER/cerb-B家族跨膜受体酪氨酸激酶成员[13],能促进上皮细胞增生,与乳腺癌[14]、 非小细胞肺癌[15]、 结直肠癌[16]等多种肿瘤密切相关。大量研究表明, EGFR基因与胃癌的发生、发展及遗传有关[17-18]。而发生在其3´UTR的SNP将影响miRNA的调控作用,新的SNP可以减弱相应miRNA对其翻译的抑制效应,增强靶基因表达或者形成新的miRNA结合位点,从而抑制靶基因的表达。
进一步扩大样本量后的病例-对照研究方法发现EGFRrs884225 C>T携带CT+TT基因型的个体与CC基因型相比,患贲门癌的风险明显增加。且根据多个生物信息学数据库预测结果,我们发现miR-31、miR-214可与 EGFR r s884225位点C等位基因结合,而EGFR rs884225位点C等位基因突变成T等位基因后,会增加更多miRNA的调控作用,形成miR-103、miR-107、miR-424、miR-486-3p、miR-497与 EGFR的新的结合位点,从而抑制了 EGFR基因的翻译过程,对贲门癌的发生具有一定的保护作用,这与我们的结果是相一致的。为了进一步探讨单体型对胃癌发生风险的影响,分别联合了 EGFR( r s884225、rs10277413)和MSH2(rs17502941、rs11125144)基因的2个3´UTR SNPs位点构建单体型,但均未发现单体型与胃癌、贲门癌、非贲门癌发病风险有关。
本次研究从消化道肿瘤相关基因3´UTR区SNPs角度探讨新发现的SNP位点是否存在与胃癌相关的遗传易感标志物。研究结果发现, EGFR基 因3´UTR的rs884225多态性位点改变可能引起miRNA对其表达调控过度导致在体内低表达,从而抑制贲门癌的发生,但具体的作用机制还需进一步实验验证。miRNA靶基因3´UTR SNPs与肿瘤关系的分子流行病学研究是目前的热点[19],通过分子流行病学的研究积累,可以对肿瘤的发生有较好的预见作用,进而有利于肿瘤的预防甚至是预后的判断。[20]
猜你喜欢
分子催化(2022年1期)2022-11-02
储能科学与技术(2022年2期)2022-02-19
感染、炎症、修复(2021年1期)2021-07-28
昆明医科大学学报(2021年3期)2021-07-22
烟草科技(2021年6期)2021-06-24
中华养生保健(2020年9期)2021-01-18
矿产勘查(2020年3期)2020-12-19
电脑知识与技术(2018年19期)2018-11-01
三联生活周刊(2017年48期)2017-11-25
医学研究杂志(2015年12期)2015-06-10
