
p53在调控DNA 损伤所致MDA-MB-231细胞死亡中的作用及其机制
2018-10-16 01:46姜朋涛胡志芳高兴春姜凤良
癌变·畸变·突变 2018年5期
姜朋涛,胡志芳,高兴春,郭 娜,张 典,姜凤良*
(西安医学院基础医学部免疫学教研室,西安 710021)
全球乳腺癌占女性癌症发病总例数的25%,占女性癌症死亡总例数的15%[1]。中国乳腺癌统计数据虽显著低于此数值,但是人口基数大,而且发病数和死亡率均逐年上升[2]。所以,乳腺癌严重威胁女性的健康,对乳腺癌发病机制的研究具有重大意义。目前乳腺癌发病机制的探索主要集中在癌细胞的存活、增殖、侵袭、转移机制等多个方面,其中基因组遗传学改变是其中一个重要的研究方向[3]。基因组遗传学改变(DNA损伤等)可以自发产生,但是更多见于各种理化因素所致,比如紫外线、电离辐射、某些药物等。DNA损伤严重时可以引起乳腺细胞的死亡,而低剂量的DNA损伤可引起乳腺细胞功能蛋白发生突变导致细胞癌变的发生。p53蛋白是一个重要的DNA损伤修复蛋白,一旦其自发或者诱发产生突变,均会引起细胞癌变[4]。所以,探索DNA损伤及损伤后修复过程中p53等相关蛋白的功能改变对于深入理解乳腺癌的发病机制有重要意义。本文拟初步研究在紫外线照射引起的乳腺癌细胞DNA损伤从而诱导细胞发生轻度死亡过程中乳腺癌细胞基因组遗传学改变相关的发病机制,这将为治疗乳腺癌寻找新靶点提供实验依据。
1 材料与方法
1.1 材料
MDA-MB-231细胞系购自上海细胞研究所;用于此细胞培养的DMEM高糖培养基和胎牛血清均购自美国Gibco公司。CCK-8细胞增殖试剂盒为日本同仁化学公司产品。剪切PARP抗体(c-PARP,1∶2 000,兔)、p21(1∶ 2 000, 兔 )、 Actin(1∶ 5 000, 鼠 )为 Cell Signaling公司产品;磷酸化p53(p-p53,1∶500,兔),p53(1∶500,鼠),磷酸化H2AX(p-H2AX,1∶500,鼠)为Abcam公司产品;NF-90(1∶1 000,鼠)为Santa Cruz公司产品。羊抗鼠-HRP(1∶2 500,115-035-003)和羊抗兔-HRP(1∶2 500,111-035-003)均为美国Jackson Immuno公司产品。
1.2 方法
1.2.1 DNA损伤诱导 本实验应用短波紫外线(UVC,简称UV,100 nm<波长<280 nm)照射诱导DNA损伤。根据紫外线输出功率计算本实验所用UVC照射箱为距离光源30 cm处时,照射1 s为1 J/m2。本实验选择照射5和20 s时剂量分别为5和20 J/m2,同时设未照射细胞为对照组。照射完成后细胞继续置于CO2培养箱中孵育相应时间后进行后续实验。
1.2.2 细胞增殖实验MDA-MB-231细胞经过不同剂量紫外线(5和20 J/m2)处理后,继续孵育24 h,按照CCK-8试剂盒说明书的步骤进行检测,每孔加入10 μL CCK-8后2 h,用全自动酶标仪读取 D(450)值,检测不同强度紫外线处理后细胞的增殖情况。
1.2.3 Western blot检测紫外线处理后不同时间磷酸化H2AX及相关蛋白质的表达水平 由于磷酸化H2AX蛋白是DNA损伤的标志物[5],通过检测其表达水平可以反映是否引起了细胞DNA损伤,同时检测与DNA损伤机制相关的p-p53、 p 53、 NF-90等蛋白。收集5 J/m2紫外线处理后不同时间点(0.5、1、2、4、8、12和24 h)的细胞,用加入蛋白酶抑制剂和磷酸酶抑制剂的RIPA裂解细胞,BCA(bicinchoninic acid)法测定细胞提取上清中的蛋白质浓度。蛋白质上样量为50 μg,配置SDS-PAGE凝胶电泳分离蛋白后转膜,5%脱脂牛奶孵育2 h后滴加一抗(详见材料部分),4 ℃孵育过夜;第2天, 滴 加二抗室温孵育1 h。常规显影、定影。应用FIJI 软件(http://fiji.sc/,基于Image J的图像分析软件)对Western blot胶片进行灰度分析,计算相应蛋白的相对表达量[6]。
1.2.4 紫外线处理后不同时间点c-PARP和p21蛋白表达的变化 检测PARP剪切(c-PARP)水平,可提示DNA损伤的修复功能受到影响的程度,这也是细胞死亡有一定增多的原因。而p21是细胞周期蛋白依赖性激酶抑制剂(cyclin-dependent kinase,CDK)家族中的重要成员,p21的表达增加,最终引起细胞周期G1阻滞的发生。为了探究紫外线引起细胞死亡的原因,我们通过Western blot实验检测了5 J/m2紫外线处理细胞不同时间点(0.5、1、2、4、8、12和24 h)后c-PARP和p21蛋白表达的变化。Western blot检测方法同1.2.3。
1.3 统计学分析
用Excel作图及进行统计学分析。计量数据以x± s表示,两组间比较采用t检验,以α =0.05为检验水准。
2 结 果
2.1 不同强度紫外线照射对MDA-MB-231细胞增殖的影响
如图1所示,与对照组(未照射组)比较,5和20 J/m2的UV C照 射MDA-MB-231细胞24 h后,5 J/m2处理组即可以引起细胞死亡增加、增殖减慢(存活率89%,P<0.05),而当细胞在20 J/m2的紫外线照射后,细胞发生明显死亡(存活率仅为22%, P<0.01)。由于本文目的为检测DNA损伤时乳腺癌细胞的保护机制,所以后续实验将使用引起细胞死亡较温和的5 J/m2处理细胞。
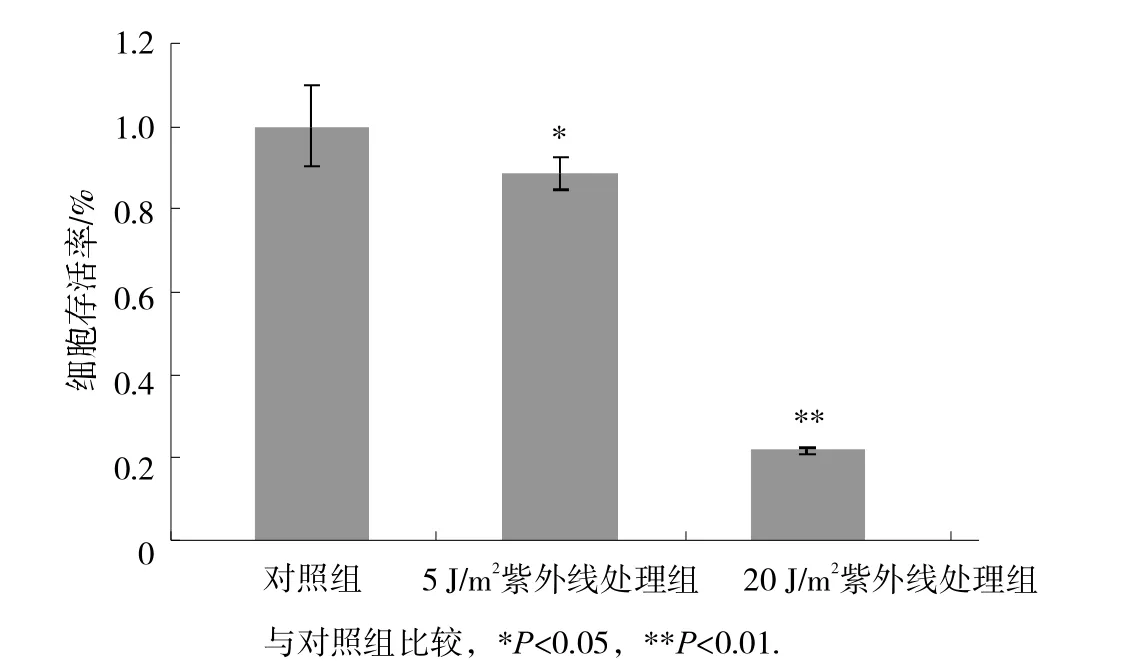
图1 不同强度紫外线处理后细胞的存活情况
2.2 紫外线照射引起p-H2AX水平的变化
图2 为Western blot蛋白免疫印迹法检测DNA损伤和细胞死亡相关蛋白c-PARP、p-p53、p53、p21、NF-90和p-H2AX结果。图3所示为p-H2AX与对照组比较的相对表达量,5 J/m2紫外线处理后0.5 h即可引起细胞DNA损伤(p-H2AX)增加(P<0.05),到2 h后,引起细胞DNA损伤增加非常明显(P<0.05),12 h时磷酸化H2AX蛋白达峰值(P<0.05),直到我们的检测终点24 h,DNA损伤较12 h时降低,但仍显著高于对照组(P<0.05)。以上结果说明紫外线处理后DNA损伤非常明显。

图2 Westernblot检 测U VC 处理不同时间后相关蛋白的表达水平
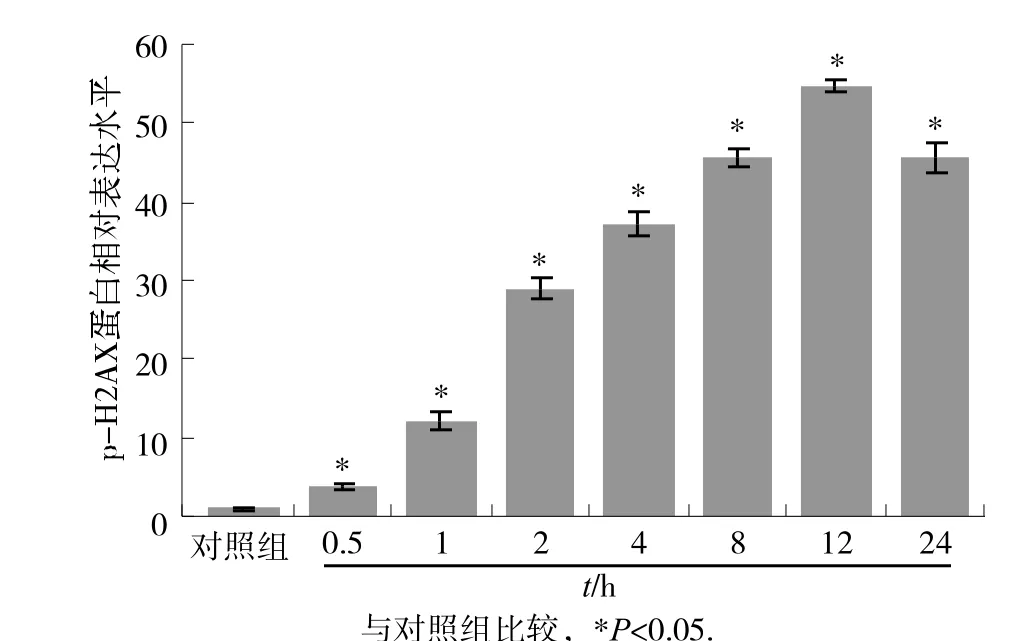
图3 UVC处 理后不同时间点 p -H2AX蛋白表达的变化
2.3 UVC通过p21-PARP通路引起细胞死亡增加
如图4A所示,与对照组比较,UVC处理8、12和24 h后c-PARP水平均明显升高(P<0.05),这提示细胞发生死亡,DNA损伤的修复功能受到影响。而p21在细胞周期调控和DNA损伤中均发挥着重要作用,它的降解将直接导致细胞DNA修复功能的损伤,与细胞死亡直接有关[7]。图4B显示在紫外线处理0.5 h后即开始出现p21的降解,并且p21的表达水平持续偏低(P<0.05),所以DNA损伤的修复功能受损与p21的快速降解密切相关。
2.4 p53参与调控DNA损伤诱导的细胞死亡
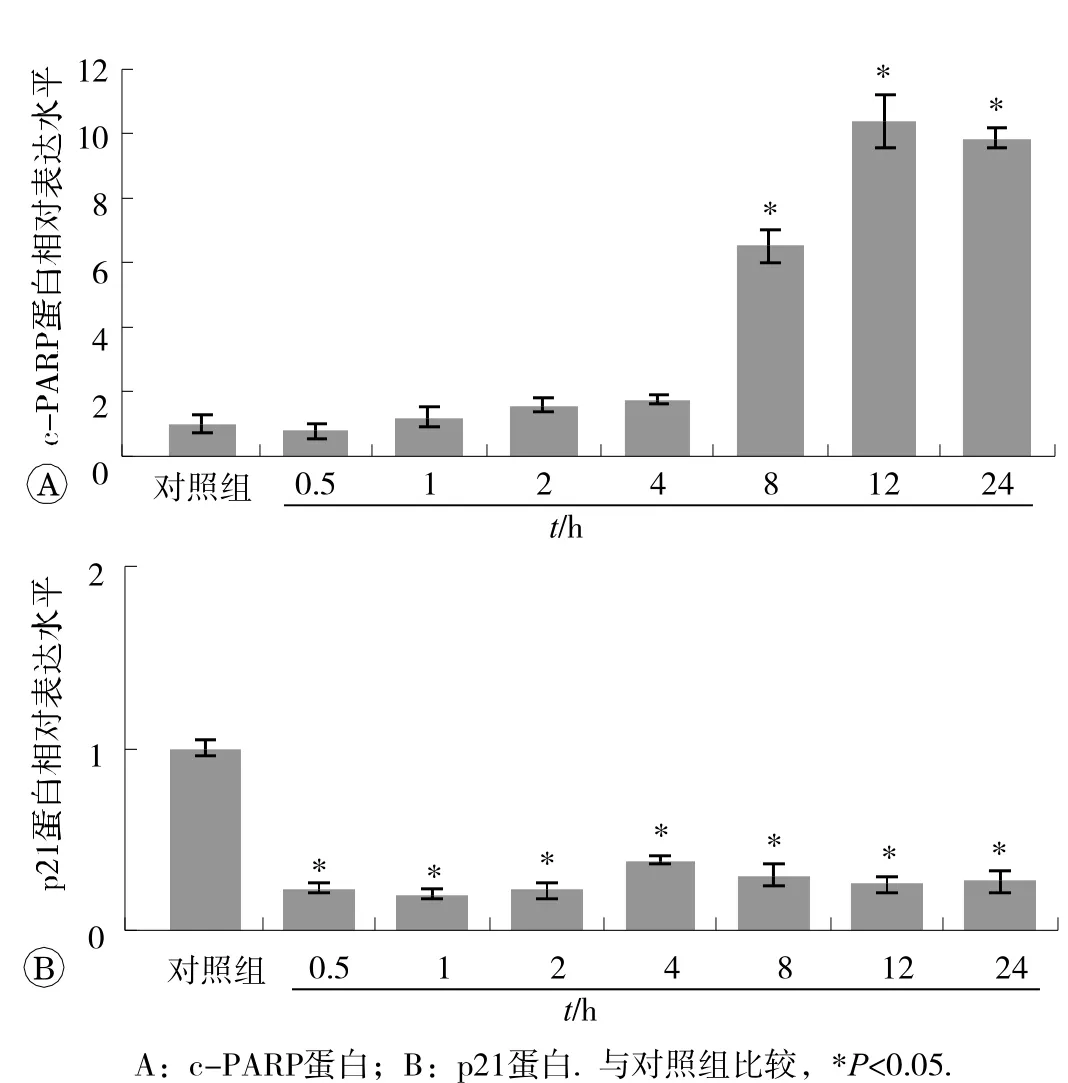
图4 UVC处 理后不同时间点 c- PARP 和 p21蛋白表达的变化
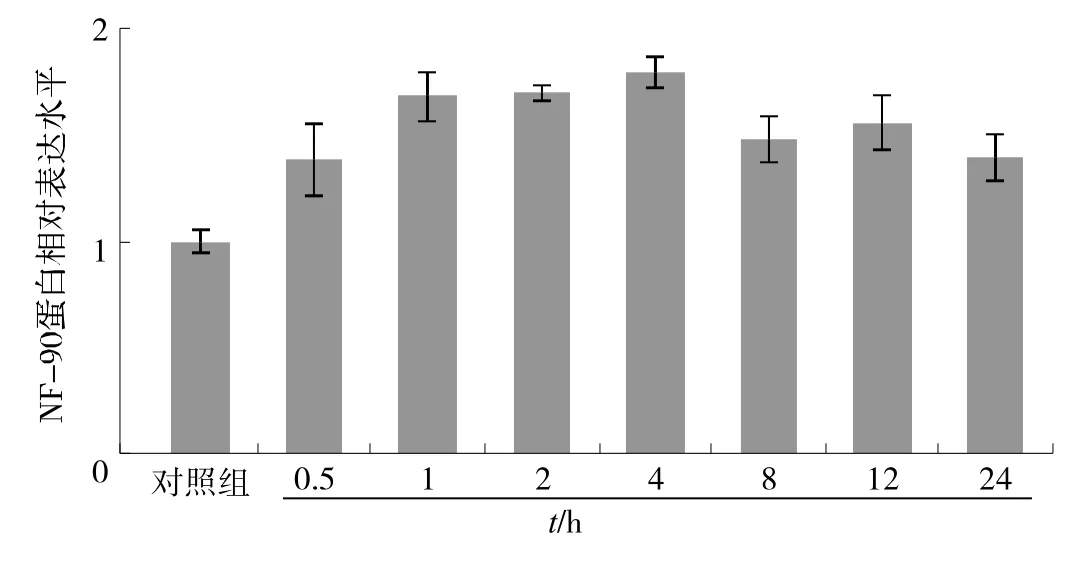
在p21蛋白持续低水平表达时,乳腺癌细胞并没有发生细胞大部分死亡。那么,什么因素参与调控这一过程呢?p21是p53重要的下游分子,同时,野生型p53可以参与对p21的调控。所以我们检测了p53的改变。如图5A所示,与DNA损伤一致,在处理早期就已经发现DNA损伤相关分子p53的磷酸化增加,在此过程中,p53并未见显著性变化(图5B)。由于核因子NF-90与NF-45形成异源二聚体在转录和翻译水平调控p53和p21的表达[8]。所以我们同时对NF-90的表达进行了检测,结果发现NF-90的表达并无明显改变,这就提示我们这一过程不依赖于NF-90蛋白量的改变(图6)。这些结果提示p53并未参与p21降解引起的细胞死亡增多,那么它的增多发挥什么作用及其具体机制仍有待我们进一步研究。

图5 UVC处理后不同时间点p-p53和p53蛋白表达的变化
3 讨 论
乳腺癌的发生、发展与多种因素导致的乳腺细胞DNA损伤修复缺失密切相关,DNA损伤引起细胞周期阻滞后细胞启动DNA修复过程,这其中包括多种功能蛋白的参与,比如ATM、BRCA-1、p53和PARP-1等[9]。在此过程中任何步骤或分子发生改变均可引起细胞癌变的发生,所以细胞对于DNA损伤后启动的保护机制需要我们进一步阐明[10]。
本文通过紫外线体外诱导乳腺癌细胞发生DNA损伤,发现在低剂量时虽然有PARP的剪切发生,这一过程与p21的快速降解密切相关,细胞发生少量死亡。p21作为一种细胞周期依赖性激酶(cyclin-dependent kinases,CDK)抑制剂,它的表达水平在多种DNA损伤刺激时均可增高,引起细胞阻滞的发生,为DNA损伤的修复提供时间。但是对于其在DNA修复中的作用仍有争议[11]。有报道发现p21 由于受到DNA损伤从而导致细胞内泛素化降解[12]。本实验观察的DNA损伤后p21快速降解(图4B)也验证了这一结果。另外,p21在细胞死亡中的作用也存在巨大争议。一般认为,p53引起p21的表达增加,可以有效抑制细胞死亡的发生,但也有相反的报道,如p21高表达的细胞在UV刺激的时候可以引起p53依赖的细胞死亡增加[13]。总之,p21由于细胞类型、细胞定位、p53状态和DNA损伤刺激的不同发挥着不同作用[14-15]。本文中我们的实验结果提示p21的快速降解导致DNA修复功能的缺失,PARP剪切增多,最终导致细胞死亡增加,但是细胞死亡并不十分明显(与高剂量相比)。那么,在这一过程中抑制细胞死亡的机制是什么呢?
p53是一个重要的抑癌基因,它在DNA损伤修复中也发挥着重要作用[16]。当DNA发生损伤时,p53磷酸化增加,从而导致与鼠双微粒体(murine double minute 2, M DM2)亲和力降低,p53被功能性释放继而激活下游DNA损伤修复蛋白,同时它募集下游分子如p21,引起细胞周期发生阻滞,这样细胞就有足够的时间对基因组进行修复,如果不能有效修复,p53又可以诱导细胞发生凋亡[17]。但是,由于p53被鉴定出是癌症中最容易发生突变的蛋白之一,突变导致上述功能的缺失,最终引起细胞癌变的发生[18]。乳腺癌相关的实体瘤或者细胞系中存在多种p53突变,在MDA-MB-231细胞中存在p53 R280K突变,此突变可以参与促进细胞存活的调控[19],并最终参与抑制DNA损伤对MDAMB-231细胞的死亡诱导作用。综上所述,结合我们的实验结果,突变的p53磷酸化增加促进其参与抑制细胞死亡的发生。
同时,我们对p53和p21的调控进行了初步研究。结果显示NF-90的表达水平并未发生明显改变。NF-90是剪切体的一个成员,含有双链RNA结合区域,它可以从细胞核转位到细胞浆结合在p21的3´UTR区从而稳定p21 mRNA[20]。在后续实验中我们将继续研究NF-90的转位对p53和p21的调控,同时我们将研究这一过程中细胞周期及其相关蛋白的作用机制。
由于癌症细胞中一些DNA修复相关分子多发生结构或者功能的改变,所以基于这些分子研究其靶向治疗药物是肿瘤靶向治疗的一个重要方向,多种相关药物已经被批准上市[21]。p53和p21既参与肿瘤抑制作用,又能通过抑制周期素依赖激酶复合物活性、协调细胞周期、DNA复制与修复之间的关系,促进肿瘤存活,所以它们在不同细胞系,不同刺激状态作用下所表现的功能改变均可以作为肿瘤药物个体化治疗的靶分子。本研究初步揭示了在乳腺癌细胞系MDA-MB-231中DNA损伤引起细胞死亡及其保护机制,进一步明确了在MDA-MB-231细胞中DNA损伤时p53和p21的改变,为基于p53或者p21的肿瘤治疗提供了实验支持。
猜你喜欢
保健医苑(2022年6期)2022-07-08
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
皮肤性病诊疗学杂志(2021年5期)2021-11-27
三农资讯半月报(2021年1期)2021-01-27
动漫星空(兴趣百科)(2020年11期)2020-11-09
天天爱科学(2020年5期)2020-09-10
时代英语·高一(2019年1期)2019-03-13
分析化学(2017年12期)2017-12-25
中国医药导报(2016年33期)2017-03-06
