
同型半胱氨酸增加阿尔兹海默症患病风险相关机制的研究进展
2018-10-16 01:46胡夏练郭锡汉马晓玲
癌变·畸变·突变 2018年5期
胡夏练,郭锡汉,马晓玲,汪 旭,3,*
( 1.云南师范大学生命科学学院,云南 昆明 650500;2.上海三誉华夏基因科技有限公司,上海 201100;3.云南师范大学生物能源持续开发与利用教育部工程研究中心,云南 昆明 650500 )
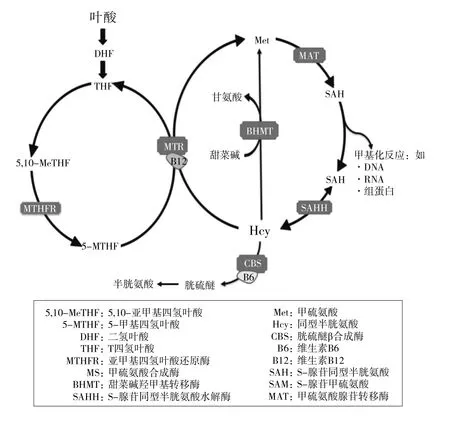
同型半胱氨酸(homocysteine,Hcy)是由甲硫氨酸经脱甲基作用产生的一种含硫的非必需氨基酸,其不参与蛋白质的合成。在一碳代谢中,甲硫氨酸腺苷转移酶(methionine adenosyltransferase,MAT)催化甲硫氨酸转变为S-腺苷甲硫氨酸(S-adenosylmethionine,SAM),SAM可作为细胞内甲基化反应的甲基供体参与甲基化,随后转变为S-腺苷同型半胱氨酸(S-adenosylhomocysteine,SAH),SAH经过可逆的 水 解产生Hcy。Hcy在甲硫氨酸合成酶(methionine synthase,MS)以维生素B12为辅酶的作用下再甲基化合成甲硫氨酸和四氢叶酸;也可在维生素B6为辅酶的胱硫醚 β合成酶(cystathionine synthase,CBS)催化下合成半胱氨酸。此外,Hcy也可与甜菜碱在甜菜碱羟甲基转移酶(betaine hydroxymethyltransferase,BHMT)的催化下合成甲硫氨酸,Hcy代谢途径见图1[1-2]。MS、CBS和BHMT等的遗传缺陷以及B6、B12等辅酶的缺乏会导致细胞内存留过量的Hcy,进入血液中,造成高同型半胱氨酸血症(HHcy)。正常人血浆Hcy浓度为5~15 μmol/L,15~30 μmol/L为轻度增高,30~100 μmol/L为中度增高,大于100 μmol/L为重度增高[3]。
阿尔兹海默症(Alzheimer's disease,AD)是一种神经退行性疾病,是痴呆症中最常见的类型,其主要表现为渐进性记忆丧失、认知衰退、学习障碍等症状,严重影响社交、职业与生活[4]。AD是遗传因素与环境因素相互作用的结果,且随着年龄的增长,个体患AD的风险增加,一些环境因素(如肥胖、抽烟和不健康的生活方式等)也会增加个体AD患病风险[5]。大量流行病学和临床研究显示,AD的发生和发展伴随着血浆Hcy的升高,HHcy使个体AD发生风险增加一倍[3-5]。现就Hcy增加AD患病风险的机制研究进展综述如下。
1 Hcy诱导β -淀粉样蛋白沉积
淀粉样前体蛋白(amyloid precursor protein,APP)是一种存在于全身组织细胞上的单次跨膜蛋白,广泛存在于脑细胞中。APP蛋白的裂解主要由 β-分泌酶(如BACE-1)和 γ-分泌酶(如早老素PS-1/PS-2)催化, β-分泌酶剪切APP形成一个可溶性细胞外N-末端(sAPPβ) ,随后 γ-分泌酶进一步剪切形成一个跨膜的C-末端(CTFβ),这种结构易于形成 β-淀粉样蛋白(β-amyloid,Aβ)[6]。Aβ具有一定神经毒性,可通过氧化应激、影响Ca2+平衡、激活caspase途径、诱导线粒体损伤等途径造成神经细胞死亡[7-8]。 Zhang等[6]在HHcy小鼠中发现Aβ水平增加的同时,APP蛋白量下降,PS-1的mRNA和蛋白水平均上调,但是 β-分泌酶的表达无显著变化,还发现APP裂解的关键位点——苏氨酸-668的磷酸化水平增加。另一方面,诸多研究证实PS-1的表达受其启动子区甲基化水平的严格调控,Hcy诱导其启动子区CpG岛的胞嘧啶甲基化水平降低,造成PS-1过表达[9],PS-1上调增加APP异常裂解为Aβ4 0和Aβ42[10],而过磷酸化的APP苏氨酸-668则增加APP与BACE-1的结合,BACE-1剪切APP形成A β[11]。此外,近期有研究发现高Hcy通过诱导细胞内SAH/SAM比(SAH/SAM ratio)增加,SAH量增多,甲基转移酶活性降低,导致5-脂氧合酶(5 LO)基因启动子区的DNA低甲基化并最终上调5LO蛋白,5LO通过激活cAMP效应元件结合蛋白(cAMP response element binding protein,CREB)来增加γ-分泌酶的表达,形成Aβ沉积[12]。另外,5LO能够促进花生四烯酸合成白三烯,白三烯是许多炎症和过敏症状的重要介质,继而诱发神经炎症[13]。
2 Hcy诱发Tau蛋白过磷酸化
Tau蛋白是1975年发现的一种神经细胞内的微管相关蛋白,由位于17q21.31的 M APT基因编码。Tau蛋白为含磷酸基蛋白,是细胞骨架的重要组成成分,其与微管结合以提高微管的稳定性,并参与神经细胞中的轴突运输,正常脑细胞中一个Tau分子含2~3个磷酸基[14]。在高水平Hcy暴露下,神经元细胞中与Tau磷酸化作用相关的去磷酸酶和磷酸激酶的表达发生改变,如蛋白磷酸酶2A(protein phosphatase 2A,PP2A)、细胞周期蛋白依赖性激酶5(cyclin-dependent kinase 5,CDK5)和糖原合成酶激酶3(glycogen synthase kinase 3,GSK-3)等。PP2A是一种Tau蛋白去磷酸化酶,Sontag等[15]的研究表明,高水平Hcy能够降低PP2A、亮氨酸羧基甲基转移酶(leucine carboxyl methyltransferase 1,LCMT1)以及PP2A/Bα全酶的甲基化水平,导致PP2A的表达下调,减弱Tau的去磷酸化,这可能是Hcy增加AD风险的一种新机制。与PP2A相反,CDK5和GSK-3都是Tau蛋白主要的磷酸化激酶,高水平Hcy能够通过激活CDK5和GSK-3通路,以增加Tau蛋白的磷酸化水平[16],Tau蛋白的高磷酸化位点主要在苏氨酸-231/丝氨酸-235和丝氨酸-396[17]。此外,Hcy可激活含半胱氨酸天冬氨酸蛋白水解酶3(caspase-3),caspase-3能够识别Tau蛋白C-末端并参与剪切Tau蛋白形成C-末端截断[18]。C-末端截断增加和Tau蛋白特异位点的高磷酸化,导致两个Tau单体易结合形成Tau二聚体,随后形成Tau低聚复合物(Tau oligomer complex 1,TOC1),Tau二聚体和TOC1不能与微管结合,导致微管稳定性下降。随后,Tau二聚体成对聚集形成螺旋纤丝,最终在神经细胞内形成神经原纤维缠结(neurofibrillary tangles,NFT),损伤神经细胞[17-18]。近年来,也有研究显示,CDK5的激活与P35和P25有关,而由Hcy诱导上调的5LO可通过提高P35和P25的表达增加CDK5的活性,导致Tau高磷酸化,且5LO对Tau蛋白的高磷酸化作用独立于其对Aβ的作用[17-19]。
3 Hcy诱发神经细胞DNA损伤
研究表明,Hcy可能通过对细胞的DNA造成损伤,导致细胞功能的紊乱[20]。在神经细胞中,DNA损伤是其凋亡的一个主要原因,而细胞凋亡诱导的神经元大量丢失可能导致神经退行性疾病的发生和发展。神经干细胞(neural stem cell,NSCs)在神经系统中具有重要作用,NSCs对DNA损伤十分敏感,如DNA链断裂、端粒长度缩短和DNA链间交联等[21]。 Wang等[21]的研究发现Hcy呈剂量依赖性地诱导NSCs的DNA链间交联,并诱发活性氧产生。Lin等[22]发现Hcy通过诱导SAH的增加,并伴DNA甲基转移酶DNMT1、DNMT3a和DNMT3b的表达和活性均降低,导致DNA甲基化异常,影响神经细胞的增殖。Currò等[24]发现Hcy诱导神经元细胞活性下降35%,拖尾细胞增加30%,DNA损伤指数增加2倍,活性氧水平增加4倍,且与DNA损伤相关的 B ax、Bcl-2、 P 53、 P 16和 P21基因均显著上调。因此,高浓度Hcy可能通过破坏DNA的二级结构,进一步诱发神经细胞的氧化损伤和遗传毒性。
此外,自由基可使DN A的碱基和脱氧核糖发生化学变化,引起碱基改变、破坏或脱落,脱氧核糖分解,形成 D NA蛋白质交联和磷酸二酯键断裂等,自由基可与核酸反应,形成许多类型的碱基修饰产物,最常见的如8-羟基-2'-脱氧鸟苷(8-hydroxy-2'-deoxyguanosine,8-OHdG)[25]。8-OHdG可作为DNA氧化损伤的重要指标,近年来,大量体内外实验证实,许多能诱导活性氧生成的外源化学物质或物理因素均可导致8-OHdG的增加。Chen等[26]的研究表明,高水平Hcy处理加重大脑皮质和海马齿状回区线粒体超微结构的损伤。此外,Hcy水平升高还显著伴随着线粒体复合物I~III酶活性的抑制以及细胞色素C的增加,同时8-OHdG水平增加。体外研究显示,使用谷胱甘肽乙酯(一种ROS抑制剂)能够抑制Hcy介导的信号转导和转录激活因子3(signal transducers and activators of transcription 3,STAT3)的过度激活,降低ROS。
4 Hcy诱导神经细胞自噬异常
自噬(autophagy)是一种由基因调控 的 对细胞内变性和错误折叠的蛋白质以及受损细胞器进行降解的过程。适度的自噬可为细胞修复和动态平衡提供营养和能量,还能降低活性氧和促凋亡蛋白的表达,但是自噬作用不足或过强均会导致细胞功能失调,如自噬性细胞死亡[27]。
近期研究表明自噬在神经细胞的活性调节中具有重要作用。雷帕霉素复合物1(mammalian target of rapamycin complex 1,mTORC1)是与细胞合成、分解和代谢紧密相关的主要激酶,mTORC1通过对自噬特征性蛋白ATG13磷酸化而抑制细胞自噬[28]。 Khayati等[29]发现高浓度Hcy导致mTORC1活性显著增加。推测Hcy通过激活mTORC1,抑制细胞内自噬作用,并造成Aβ和Tau等异常蛋白的积累[29-30]。此外,Notch信号通路在细胞分化中具有重要作用,已有研究表明Notch与自噬的信号调控有关[31]。发状分裂相关增强子1(hairy enhancer of split 1/5,hes1/5)是Notch1受体下游信号分子,介导神经元再生,其缺乏会引起严重的神经损伤。Zhang等[32]在高水平Hcy小鼠中发现hes1和hes5的表达均降低,线粒体空泡化、神经元凋亡增加、自噬体增多,推测Hcy可能诱导神经细胞过度自噬和凋亡。另外,有研究表明PS-1/PS-2在介导自噬过程中也具有重要意义[33],PS-1表达下调导致自噬不足,而高水平Hcy能诱导神经细胞中PS-1过表达引起神经细胞自噬过度[34]。 Zhao等[35]的研究表明,Hcy可通过诱导神经细胞中8-OHdG含量的增加,造成神经细胞氧化损伤,且与自噬相关的蛋白轻链 3( light chain 3,LC3)和beclin-1表达增加,引起自噬过度。
5 结语
Hcy是一碳单位代谢中的重要中间产物,是AD风险增加的主要因素之一,维生素B12、B6和叶酸的缺乏,甲硫氨酸的过量摄入等都能够导致体内Hcy浓度升高。HHcy通过诱发神经细胞外Aβ沉积、细胞内Tau蛋白过磷酸化、神经元DNA损伤、神经炎症、细胞凋亡和自噬异常等在AD发展过程中发挥关键作用。基于年龄(以65岁为界限)可将AD分为两类:早发性阿尔兹海默症(early-onset Alzheimer’s disease,EOAD)和晚发性阿尔兹海默症(late-onset Alzheimer’s disease,LOAD)[4,36]。高浓度Hcy可通过对基因表达的影响,诱发一系列的细胞应答及信号通路。有研究显示,Hcy可能通过影响相关基因如APP、 P S-1、 P S-2等的遗传变异,增加EOAD的发生和发展,造成AD的发病年龄提早化[37]。此外,近年来有研究用孟德尔随机化(mendelian randomization,MR)从遗传学和基因角度系统评价风险因素与AD之间的因果关系,认为端粒长度(telomere length,TL)缩短与AD风险的增加密切相关[38]。但是,Hcy对神经细胞TL的影响鲜有报道,因此,设立较好的MR研究有助于确定AD风险与遗传因素之间的关系。虽然Hcy诱导AD风险增加的分子机制仍存在许多未知,需要更为深入的体内外研究以及动物模型研究来探讨,但是Hcy对AD发病机制的进一步揭示可以为AD等神经退行性疾病的防治提供新的策略和方向。
猜你喜欢
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
科学24小时(2022年2期)2022-01-26
昆明医科大学学报(2021年10期)2021-12-02
家庭医药(2021年7期)2021-07-23
昆明医科大学学报(2021年2期)2021-03-29
昆明医科大学学报(2021年1期)2021-02-07
中华养生保健(2020年4期)2020-11-16
祝您健康(2020年5期)2020-05-14
分析化学(2017年12期)2017-12-25
