
生物类似药分析相似性评价的相关统计学方法考量*
2018-07-16 10:06吴克坚赵清波李婵娟潘海涛夏结来
中国卫生统计 2018年3期
吴克坚 赵清波 李婵娟 王 陵 李 晨 潘海涛 夏结来△
【提 要】 目的 在生物类似药的质量比对研究中,为评价候选药与参照药的分析相似性,FDA建议对3个不同层级的关键质量属性采用不同的评价方法。本文建立第1层级的关键质量属性分析相似性评价的步骤及方法,重点讨论如何确定候选药与参照药的样本量nT,nR。方法 利用判断方差齐性的F检验介绍方差相似性检验假设及统计量的建立;利用高个体差异药物的生物等效性评价方法及双单侧检验的性质等建立分析相似性检验假设及统计量,并由推导得到的检验效能的精确公式确定样本量。结果 在分析相似性评价过程中将nR控制在[nT,1.5nT]内,根据数值模拟得到不同参数组合的样本量,提出了确定nT,nR的策略,在此基础上给出了评价生物类似药分析相似性的具体步骤和方法。结论 本文提出的样本量确定策略及评价分析相似性的步骤与方法可应用到实际的分析相似性研究中;同时由推导得到的检验效能精确公式还可获得其他参数组合的样本量。
自上世纪80年代开始,生物药陆续上市,比如生长激素、细胞因子、重组单克隆抗体等,它们在一些代谢性疾病、肿瘤、自身免疫性疾病等领域中显示出了明显的临床价值。近年来,随着原研生物药的专利到期或即将到期,越来越多的药企加入到生物类似药的研发行列中,为患者提供与原研药在质量、安全性和有效性上高度相似的生物类似药,满足患者的用药需求,降低医疗成本[1];比如美国生物制药公司辉瑞(Pfizer)和韩国生物制品制造商Celltrion共同开发的英夫利昔单抗的生物类似药Inflectra比其品牌药Remicade价格低15%,并有文献指出在一项调查研究中的患者使用该生物类似药每年可节约医疗成本约19%[2]。
生物类似药相对分子质量较大、结构复杂,具有独特的免疫原性和活性的安全隐患,给研发和监管带来了相当大的挑战;并且它不同于小分子化学仿制药的有机合成,非“生物仿制药”,故传统的证明仿制药与参照药生物等效的方法并不能直接适用于生物类似药[3]。因此,许多监管机构都发布了相关的指南文件或技术规范,如欧洲药品管理局(EMA)、美国食品和药物管理局(FDA)、世界卫生组织(WHO)等[4-6]。我国国家食品药品监督管理总局(CFDA)也于2015年3月发布了《生物类似药研发与评价技术指导原则(试行)》,其中明确了生物类似药的定义,提出了生物类似药研发和评价的基本原则,包括药学、非临床及临床研究和评价等内容[7]。然而随着生物制药公司对研发生物类似药的兴趣日渐浓厚,生物类似药的品种越来越多,上述指导原则或技术规范显得不够详细,有些具体的要求及细节未能面面俱到。因此,国内外一些学者讨论了很多具体的统计方法,比如分析相似性的评价方法、样本量(批次)估计、如何全面获得原研药质量参数等[8-10];再如,讨论证明疗效高度相似的统计方法[11]以及生物类似药贝叶斯自适应的临床试验设计方法[12]等。
FDA在发布的阐述生物类似药审批流程的3个指南文件草案表示,对注册申请生物类似药的全部提交资料采用“证据链完备性(totality-of-evidence)”的方法来审查,并鼓励用“逐步递进法(stepwise)”来分阶段证明其与参照药的相似性,并在后续的研发步骤中设法解决两者的差异。证据链完备性的方法形如“金字塔”,处于第1层的是质量比对研究,处于最高层的是临床相似性研究[13]。质量比对研究是指采用适合的、最先进的分析检测方法对候选药及参照药开展全面的理化性质、结构功能、生物活性、杂质和纯度等方面的研究,然后进行头对头比较以说明候选药与参照药在各质量参数上是否相似。因此逐步递进法始于质量参数的分析相似性研究。例如FDA批准山德士公司(Sandoz)的生物类似药Zarxio(参照药为安进药物Neupogen);再如FDA批准的首个阿达木单抗生物类似药Amjevita(ABP501)(参照药为修美乐Humira),由美国生物制药公司安进(Amgen)研发,都对它们与其参照药在一级结构、高级结构、纯度、糖基化、生物活性等层面进行了系统、充分的分析相似性评价[14-15]。
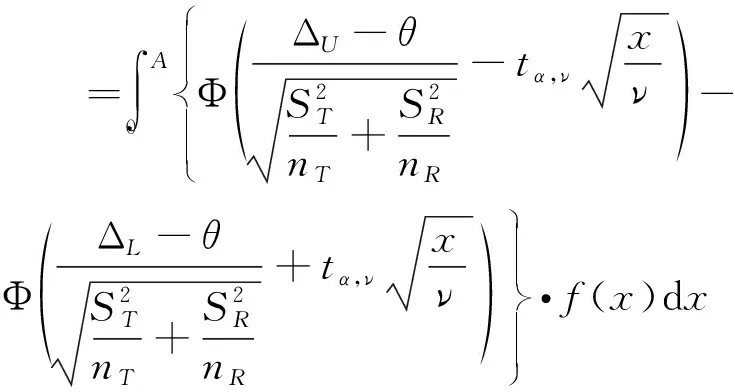
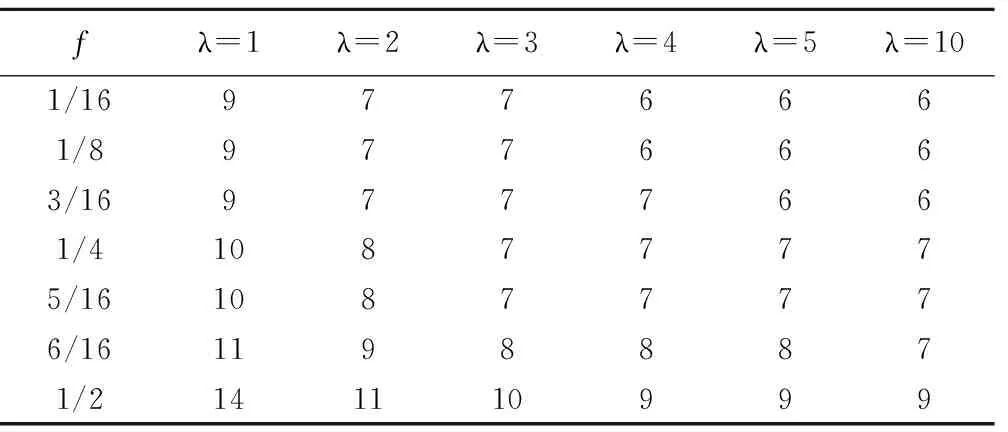
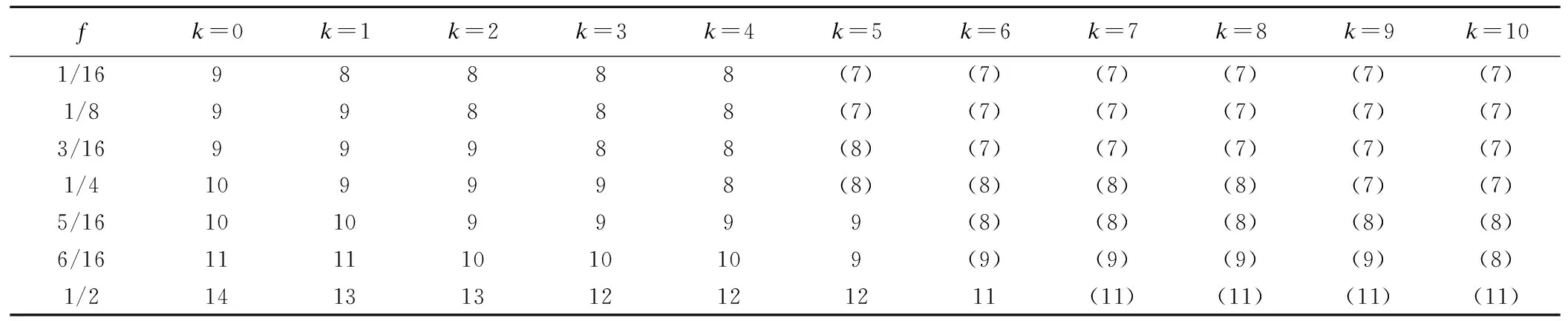
质量比对研究是生物类似药研发的必经之路,也是简化非临床和临床研究的基础。Chow,Tsong与Dong等人重点讨论了此阶段分析相似性评价方法、界值确定等问题,并给出了检验效能1-β及样本量的计算方法[10,16-19]。生物类似药成分和结构比较复杂,同一批次的生物药在不同的环境和条件下即使采用相同原理的分析检测方法可能得到不同的结果;或者不同批次生物药的结构、理化性质常存在差异,因此有必要将候选药与参照药的变异都考虑进来。然而上述文献未纳入这个重要的因素,没有经过统计分析假定候选药与参照药的总体方差相等,这样可能会导致得到的相似性结论不准确。同时根据我们在药企实地调研的结果,由于费用成本等问题,在实际的质量比对研究阶段候选药的生产批次和参照药的获取批次都非常有限;且常常需要较多批次的参照药以全面了解其理化及生物学性质;这时会造成两者样本量的严重不对等。Dong等人讨论了这一情况,指出当NR>1.5NT时需要调整样本量(NR,NT分别表示实际可获得的参照药与候选药的批次),给出了具体的调整方法[19]。然而当NT 本文大致结构如下:首先介绍基于质量属性分级体系的统计分析方法;其次讨论评估第1层级的关键质量属性的分析相似性方法,通过推导得出的检验效能精确公式计算样本量;最后数值模拟各种参数组合下的样本量方案,给出包括样本量确定策略在内的分析相似性评价方法的主要步骤,并结合实例分析进行说明。 通过对生物药结构、理化性质等方面的分析测定,我们可以得到一系列关键质量属性(critical quality attributes,CQAs),它们以不同的数据类型反映生物药的生物学活性、体内药动学、体内药效、免疫原性等特征,或多或少地影响着临床效果。由于CQAs数量很多,所以FDA建议根据其关键程度及作用机理,或者通盘考虑、验证研究,建立3个层级的CQAs分级体系,然后使用不同的方法来进行分析相似性评价。 如果某些CQAs直接影响临床效果、产品质量及安全性,或直接表明首要作用机制,我们将其归到第1层级。比如生物类似药Zarxio第1层级的CQAs有4个:氨基酸序列、效价、靶点结合力和蛋白浓度[16]。第1层级的CQAs是分析相似性研究中的重点,是判断质量可比性的重要基础,采用类似于等效检验(equivalence test)的评价方法。 由于第1层级的CQAs直接影响临床效果,在分析相似性中最为重要,要求的统计方法最为严格,所以本文主要讨论第1层级CQAs分析相似性的评价步骤与方法。首先我们进行总体方差相似性检验,经检验若认为候选药与参照药在该CQA的总体方差是相似的,则接着进行分析相似性检验。具体如下: (1) 其中(ωL,ωU)是方差相似性界值,界值有很多选择,如(0.67,1.5)、(0.5,1.5)等。在这里我们将界值设置得宽泛一些,为(0.5,2)。方差相似性检验假设(1)可以分解为双单侧检验(two one-sided test,TOST): (2) (3) 利用判断两个方差是否相等的F检验,双单侧检验问题(2)、(3)的拒绝域分别为 (4) H0:μT-μR≤ΔLorμT-μR≥ΔUvs. Ha:ΔL<μT-μR<ΔU (5) 其中(ΔL,ΔU)是分析相似性界值,FDA建议设置ΔU=-ΔL=c·σR,式中σR表示通过对多个批次参照药的研究或其他方法得到的该CQA的总体标准差。根据高个体差异药物的生物等效性评价方法——标化均值生物等效性(scaled average BE,SABE)的等效界值的设置,系数c通常设置为1.5[18]。 分析相似性检验假设(5)可以分解为: H01:μT-μR≤ΔLvs.Ha1:μT-μR>ΔL (6) H02:μT-μR≥ΔUvs.Ha2:μT-μR<ΔU (7) (8) 利用双单侧检验的性质[21],双单侧检验问题(6)、(7)的拒绝域分别为 (9) 由于生物药本身的复杂性,两种来源的生物药在结构或组成方面几乎不可能完全一致,故候选药与参照药的真实差异μT-μR≠0。根据拒绝域,我们可以得到双单侧检验(6)、(7)的检验效能函数为: χ2(ni-1)(i=T,R), 则 (10) 在前述方法部分,为了评价第1层级CQAs的分析相似性,我们认为首先应该讨论候选药与参照药的总体方差是否相似;若两者总体方差相似,接着根据推导得出的检验效能精确公式计算参数给定时的样本量,进而由样本分析值得出的拒绝域或者置信区间做出推断结论。 按照FDA的建议,令分析相似性界值ΔU=-ΔL=c·σR。为简单起见,我们假设候选药与参照药的真实差异μT-μR也与σR成比例,即μT-μR=θ=f·σR,比例常数f< (11) 表1 不同f值(μT-μR=f·σR)、λ值(nR=λ·nT)对应的nT 基于上述讨论,两组样本量的倍数关系nR=λ·nT不太适用,我们假设两组样本量关系为nR=nT+k,其中k=0,1,2,…,「0.5×nT⎤,符号“「⎤”表示向上取整数。表2给出了当α=0.05,1-β=0.8及分析相似性界值ΔU=-ΔL=1.5×σR时,不同k值、不同f值对应的样本量nT,表中带“()”的数据表示不满足条件nT≤nR≤1.5nT的样本量nT。如表2第2行k=5列对应的数据“(7)”表示所得的样本量为nT=7,nR=12不满足nT≤nR≤1.5nT。 表2 不同k值(nR=nT+k)、f值(μT-μR=f·σR)对应的nT 综上,我们可以得出评价第1层级的某CQA的分析相似性的步骤为: (1)根据样本分析值由拒绝域公式(4)判断候选药与参照药在该CQA的方差是否相似。若两者在该CQA的总体方差相似,则进行分析相似性评价; (2)通过预试验或专家预判,确定分析相似性界值ΔU=-ΔL=c×σR及真实差异μT-μR=f×σR中的c和f,如果实际获得的候选药与参照药批次关系为NR>NT,则由类似于表2的结果确定进行分析相似性评价的候选药批次nT和参照药批次nR; (3)采用所有参照药的批次数据估计该CQA的总体标准差σR; (4)从所有参照药的批次NR中随机抽取样本数据nR,根据拒绝域公式(9)判断候选药与参照药该CQA是否具有分析相似性; (5)重复第(4)步105次,统计得到两组该CQA的“分析相似性”结论的次数。若频率大于95%则可作出推断,候选药与参照药在该CQA具有分析相似性。 已知进行分析测定的参照药与候选药的实际批次分别是NR=52,NT=8。某CQA具体的样本分析值分别为: XR=(10.9,9.7,10.2,9.4,10.2,10.1,10.0,9.8,9.2,9.1,9.0,9.2,9.8,9.7,8.9,10.2,10.1,9.6,9.3,9.1,9.3,9.8,9.6,8.5,9.0,8.9,9.8,8.7,8.3,9.3,9.2,9.4,8.8,10.5,10.6,9.3,9.4,10.0,8.8,9.2,9.0,10.3,9.0,8.3,8.6,8.6,8.4,8.5,8.7,8.7,8.3,8.3);XT=(9.8,9.0,9.2,9.0,10.2,8.6,9.4,9.0)。 本文通过推导的检验效能精确公式,数值模拟给出了不同参数k,f(nT=nR+k、μT-μR=f×σR)对应的样本量方案,得出了评价某第1层级CQAs的分析相似性的具体步骤和方法,可行性强,对药企研究人员有很好的实践指导意义。也就是说,如果对于不同的生物制品,药企经过预实验后得到系数f、c的估计值,则可以通过本文提出的样本量选取策略,确定候选药与参照药的样本量,根据本文提出的分析相似性评价步骤,作出两者是否具有分析相似性的推断结论。方 法
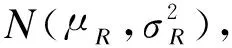
1.方差相似性检验
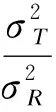
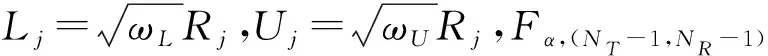
2.分析相似性检验
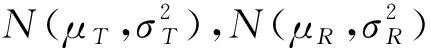
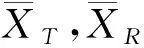

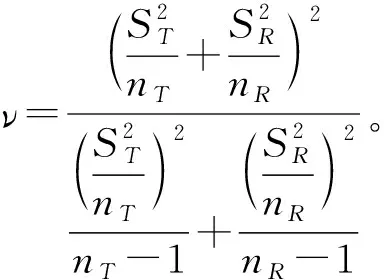


模拟与结果
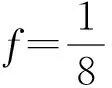
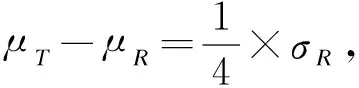
实 例
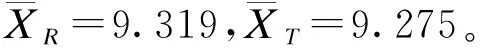

讨 论
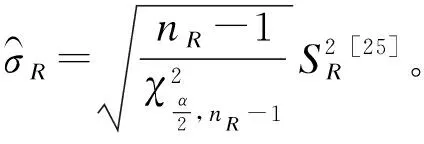 猜你喜欢
数学物理学报(2022年5期)2022-10-09内蒙古统计(2021年4期)2021-12-06河北画报(2020年8期)2020-10-27中国卫生统计(2019年3期)2019-07-10中国卫生统计(2019年3期)2019-07-10中国妇幼健康研究(2017年12期)2018-01-31数学学习与研究(2018年20期)2018-01-07汽车与安全(2016年5期)2016-12-01浙江大学学报(工学版)(2016年2期)2016-06-05俄罗斯问题研究(2013年1期)2013-03-11
猜你喜欢
数学物理学报(2022年5期)2022-10-09内蒙古统计(2021年4期)2021-12-06河北画报(2020年8期)2020-10-27中国卫生统计(2019年3期)2019-07-10中国卫生统计(2019年3期)2019-07-10中国妇幼健康研究(2017年12期)2018-01-31数学学习与研究(2018年20期)2018-01-07汽车与安全(2016年5期)2016-12-01浙江大学学报(工学版)(2016年2期)2016-06-05俄罗斯问题研究(2013年1期)2013-03-11
