
篮子试验在抗肿瘤靶向药物研发中的应用现状*
2018-07-16 12:23:16第四军医大学卫生统计学教研室710032
中国卫生统计 2018年3期
第四军医大学卫生统计学教研室(710032)
胡海霞 王 陵 李婵娟 夏结来△
随着基因组学和分子生物技术的进步,人们对肿瘤的认识越来越深入,对肿瘤的分类不再局限于组织病理学分型,也开始关注引起肿瘤发生的基因或分子改变,同时在肿瘤治疗方面也开始从传统的针对肿瘤组织部位的治疗模式转向针对肿瘤特异性遗传变异的治疗模式,即靶向治疗模式。早在20世纪80年代,全世界范围内就开始研究分子靶向药物,但很多药物的临床试验遭受了失败,其原因在于采用传统的临床试验方法进行药物研发,而没有在“正确的人群”中进行试验[1-2],即没有在有特定靶向分子变异的人群中进行试验。长春瑞滨与吉非替尼在未经选择的老龄非小细胞肺癌(NSCLC)患者中疗效并无差异,而在EGFR FISH阳性的NSCLC患者中,两者的疗效差别有统计学意义,长春瑞滨效果优于吉非替尼[3],如果没有在EGFR FISH阳性的亚人群中进行比较,一个有效药物很可能就会被埋没。
随着新一代测序技术(next generation sequencing,NGS)的推广,针对特异性变异进行肿瘤靶向治疗的模式越来越成为药物研发的热门方向,对适合靶向药物研发的新的临床试验设计也显出了越来越迫切的需求,篮子试验(basket trial)设计应运而生[4-5]。鉴于国内尚无对篮子试验全面的介绍,本综述欲从篮子试验的提出、设计及其目前在抗肿瘤靶向药物研发的应用方面进行较详细的阐述。
由于在同一组织类型的肿瘤中具有某种特异性变异的患者仅占很少一部分,为靶向药物临床试验招募研究对象带来挑战[6],而同样的基因或分子变异可能出现在不同组织类型的肿瘤中,因此篮子试验将具有同种基因或分子变异的不同肿瘤组织类型的患者全部纳入试验[7],以探索对试验靶向药物敏感的肿瘤组织学类型。由于在一个临床试验框架下,包括了多种肿瘤类型患者,可以达到多个独立临床试验的目的,因此被形象地称为“篮子”试验。其设计示意图如图1所示。
据查阅到的资料显示,2014年在美国癌症研究学会(American Association for Cancer Research,AACR)癌症进展中,篮子试验作为精准癌医学的创新性临床试验代表之一,被正式提出。在同年美国临床肿瘤学会(American Society of Clinical Oncology,ASCO)的会议上,篮子试验也被多次提到[8]。但篮子试验思想的应用远早于其方法的正式提出,2008年伊马替尼的Ⅱ期开放单臂试验[9]将所有含酪氨酸激酶相关变异的肿瘤患者全部纳入试验,而不关注其肿瘤的组织学来源,试验共纳入了40种肿瘤类型的患者,对不同肿瘤患者服用伊马替尼的效果分别进行评价,其设计思想同篮子试验如出一辙。直到2014年篮子试验被正式提出后,学术界对篮子试验的研究及应用篮子试验设计的临床试验才逐步多了起来。
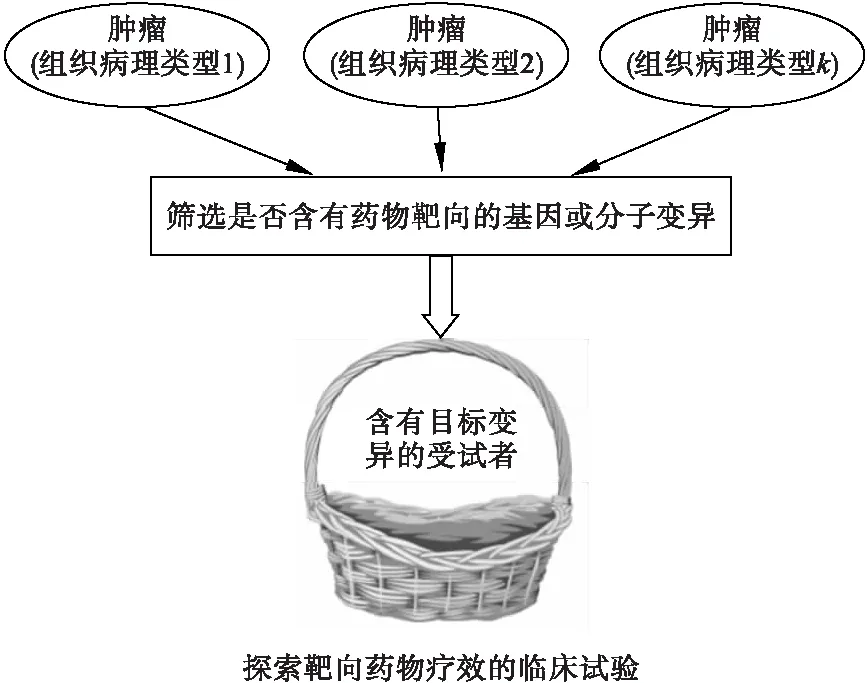
图1 篮子试验设计示意图
目前根据药物的作用机制和肿瘤患者的分子变异选择情况,有以下几种类型的篮子试验[10]:(1)研究一个药物对同一分子变异的多种肿瘤类型的疗效,典型的例子就是威罗菲尼治疗含有BRAF V600变异的非黑色素瘤恶性肿瘤的篮子试验[11]。(2)研究一个药物对少数几种分子变异的多种肿瘤类型的疗效,如正在进行的临床试验CREATE,研究克唑替尼在含有ALK和/或MET通路变异的多种肿瘤中的疗效[12]。(3)研究多个靶向药物在多种分子变异的多种肿瘤类型中的疗效,如正在进行的大型精准医学研究MATCH[13]。篮子试验形式多样,适用性广,没有固定的模式[14],这一特点十分有利于其在靶向治疗时代的推广应用。
探索性篮子试验设计
传统的探索性Ⅱ期篮子试验设计将有相同目标基因异常变异的肿瘤患者按照其肿瘤不同组织部位和分型分为不同的组,根据不同肿瘤组患者对靶向药物的反应率(response rate,RR)判断对药物敏感的肿瘤组织类型,为以后的验证性临床试验提供依据[9,15],或将篮子试验思想与Simon最佳两阶段Ⅱ期临床试验设计方法[16]相结合,用最少的样本量识别对试验药物敏感的肿瘤类型[11]。
在Simon最佳两阶段Ⅱ期临床试验设计与篮子试验结合的基础上,Kristen Cunanan等人[17]提出将适应性设计和异质性检验融入篮子试验的设计方法。即在期中分析时对k个不同肿瘤类型组的反应率进行多组χ2检验,计算其同质性系数λ(λ∈(0,1)),λ值大于预设界值时认为组间不同质,此时分别对每组反应率进行单样本率检验,与终止试验界值Tsk进行比较,以决定是否终止该组试验,未终止的试验组进入第二阶段试验,结束后再分组分别进行统计分析,做出最后疗效的判断;若其λ值小于预设界值则表示组间同质,此时将k组合并,进行单样本率检验,与终止试验界值TC进行比较,若不能终止试验,则所有组均进入第二阶段试验,结束后合并分析判断试验药物是否对所有肿瘤组织类型均有效。其流程图如图2所示。
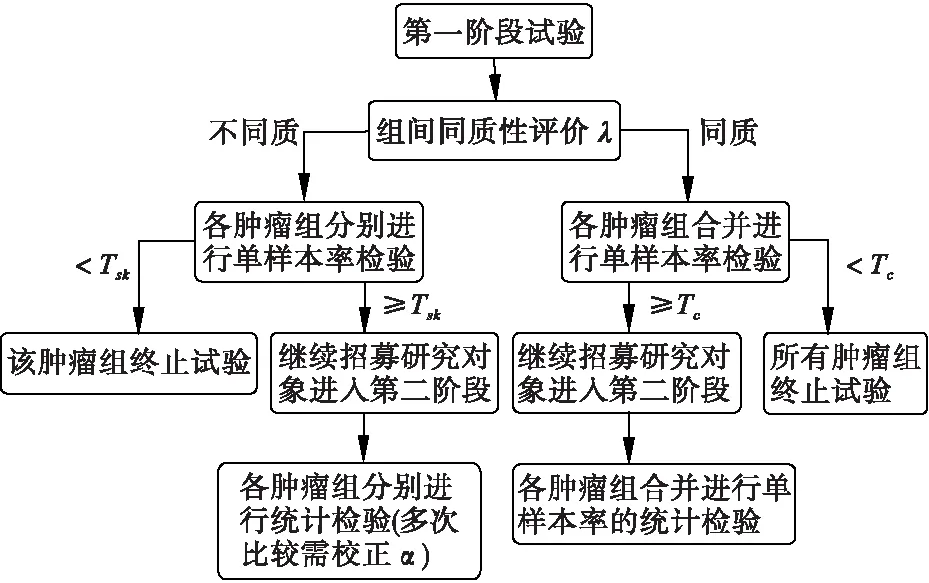
图2 异质性检验两阶段篮子试验设计流程图
这种设计与Simon最佳两阶段设计的篮子试验相比,当药物对所有或绝大多数肿瘤组有效时,可以用较小的样本量达到较大的检验效能,当药物仅对少数组织类型的肿瘤有效时,可能会损失一定的检验效能,但从总体看,可以节约样本量,提高各肿瘤组的检验效能,缩短研究时间,是值得尝试的试验设计。
Richard Simon[18]提出的贝叶斯篮子试验设计将贝叶斯思想融入Ⅱ期篮子试验,试验设计阶段需先确定研究药物对各个肿瘤组疗效的先验概率Pr[p=x],其中x为k个肿瘤组各自的效应量,仅可取Plo(认为无效的效应量)和Phi(认为有效的效应量)两个值之一,这两个值均需根据临床专业知识预先设定,p表示各肿瘤组出现该效应值的概率(p1,p2,…,pk)。此外还需设定组间效应完全关联的概率λ和各组均有效的概率γ。根据试验期中分析的结果,计算相应的后验概率并将计算所得的后验概率与预先设定的有效终止界值T进行比较,若Pi>T,认为第i肿瘤组有效,终止该组。若Pi<1-T,认为第i组无效,也终止试验,否则,继续进行试验,直到所有组均被终止或达到预设的最大样本量N。其流程图如图3所示。
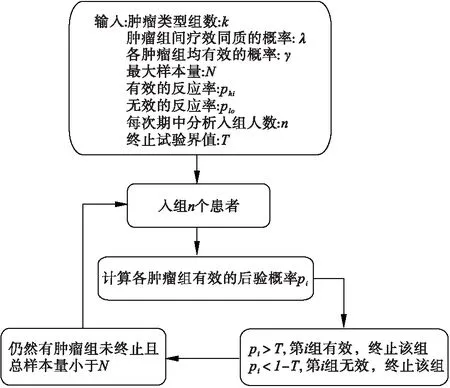
图3 贝叶斯篮子试验设计流程图
贝叶斯篮子试验与传统探索性篮子试验相比,不仅在肿瘤组间具有同质性时,可以实现组间信息共享,从而减少样本量,也可以利用试验设计者或临床专家的经验估计,以先验概率的形式体现到试验中,进一步提高检验效能。但两者均没有设立对照组,提供的临床证据较弱。
验证性篮子试验设计
篮子试验同时研究具有相同分子变异的多种肿瘤类型,由于异质性的存在,具有相同变异的不同部位的肿瘤对同一靶向药物的反应不一定相同,而无效肿瘤类型组的存在可能导致整个篮子试验的失败,因此篮子试验目前主要用于探索性试验[9]和有突破性疗效的靶向药物的验证性试验[11,19]。为了克服肿瘤异质性的问题,将验证性篮子试验推广应用到所有靶向药物的Ⅲ期临床试验,不同的试验设计类型相继被提出。在此主要介绍两阶段适应性设计的验证性篮子试验和成组序贯富集设计的验证性篮子试验。
1. 两阶段适应性设计的验证性篮子试验
2016年,Chen Cong等人[20]提出两阶段适应性设计的验证性篮子试验,并研究了其Ⅰ类错误控制方法和样本量计算公式。其思想是k个有相同分子变异的不同组织学类型的肿瘤组分别自设对照,在t点(t表示观察到的信息在总信息中的比值[21],t点样本量n=Nt,t∈(0,1))进行期中分析,第一阶段试验结果(各组标准化计分检验统计量Z1i,i=1,2,…,k)显示试验药物对某组织类型的肿瘤很有可能无治疗效果(Z1i

图4 两阶段适应性设计的验证性篮子试验流程图

图5 成组序贯富集设计的验证性篮子试验流程图
在此基础上,Beckman[22]提出了k个组共用对照组或采用外部对照的单臂试验来进一步减少篮子试验的样本量。在期中分析时,建议采用结合外部数据来剔除无效组,以减小对合并分析检验水准α*的“惩罚”,即采用“Ⅱ期+”方法[23]。在Ⅱ期探索性试验阶段结束后,对进入验证性阶段的组继续随访,以获得其最终结局数据,作为两阶段适应性设计的验证性试验期中分析剔除无效组的外部数据支持。此后,Li Wen 等人[24]提出了两阶段适应性设计篮子试验最终合并分析治疗效应的点估计值计算公式。
2.成组序贯富集设计的验证性篮子试验
篮子试验中试验药物治疗无效的肿瘤类型可能会导致整个篮子试验出现阴性结果,同理,试验中治疗效果非常好的肿瘤类型可能会导致最终合并分析时高估药物对其余肿瘤类型的疗效。因此,Yuan等人[25]提出了成组序贯富集设计的篮子试验,主张根据第一阶段试验结果同时剔除可能无效肿瘤组和疗效非常好的肿瘤组,剩余的组进行合并序贯分析,直到得到有统计学意义的结果。
以各肿瘤类型组分别自设对照为例,第一阶段试验结束后,t1点进行期中分析,当肿瘤组标准化计分检验统计量小于检验下限界值时,接受H10(第一阶段原假设),即认为试验药物对该肿瘤类型无效(IA={i:Z1i
通过模拟,研究指出最佳t1为0.2~0.5,同时剔除无效肿瘤组和效果很好的肿瘤组相比只剔除无效组可以很好地降低总错分率(将无效肿瘤组判定为有效或将有效肿瘤组判定为无效)。两阶段适应性设计和成组序贯富集设计的验证性篮子试验都能很好地控制Ⅰ类错误,用尽量少的样本量达到试验目的。但是两种方法在设计、操作上相对传统的平行组设计均较复杂,在实际操作中给临床试验者和统计分析人员带来较大难度。
篮子试验的应用
篮子试验是为了满足靶向药物研发应运而生的产物,由于其只关注是否有相应的分子变异,而不限制肿瘤组织学来源和分型,为发病率很低、临床试验困难的罕见肿瘤的药物研发提供了可能[20];对于药物研发企业,篮子试验用较少的样本量同时研究一种药物对多种肿瘤类型的治疗效果,可以显著缩短药物上市的时间,减少研发成本;对于FDA、CFDA等药品审批部门,Ⅱ期篮子试验结果可以为其评价申请药物的疗效、安全性提供依据,相信Ⅲ期篮子试验也将逐渐发挥其优势性。
但是,篮子试验作为一个新兴的临床试验设计方法,在实际操作过程中,仍然有一些局限及需要注意的地方。首先,由于肿瘤自身的异质性,同一肿瘤实体内部不同部位活检得到的“驱动基因”不同,也就可能导致目标变异并不是主要变异的肿瘤被纳入到试验中,降低试验的检验效能。另外,由于基因突变的不稳定性,同一肿瘤在接受治疗后可能发生继发突变,原先的“乘客基因”变为“驱动基因”[6],或者新的突变使得含有目标变异的肿瘤对相应的靶向治疗不再敏感,如含有BRAF V600变异的结直肠癌由于继发组织特异性的EGFR反馈环路激活导致对威罗菲尼治疗不再敏感[26],因此在篮子试验招募研究对象时,需要对患者肿瘤进行新的活检检测,而不能依赖其初次诊断或初次活检的结果进行判断[6]。其次,具有相同分子变异的不同组织部位的肿瘤异质性也较大,因此篮子试验仍然有很大的失败风险,伊马替尼Ⅱ期篮子试验入组40种肿瘤类型的病人,最终只有4种得到FDA的审批[9],因此做好研究肿瘤类型的初筛,尽可能少纳入无效肿瘤类型,可以有效提高篮子试验的成功率,推荐尽可能在传统试验已经证明有效的靶向药物和靶点的基础上开展篮子试验[22]。在Ⅲ期适应性设计的篮子试验期中分析时,由于很多生存终点(如总生存率overall survival,OS)需要的随访时间长,信息量大,不能很快得出结论,影响后期患者入组,因此有专家建议期中分析时采用中间终点(如无进展生存期progression-free survival, PFS)来代替最终终点[20,22,24],中间终点即时性好、更灵敏,可以对试验进行及时调整[24],但是需要对中间终点和最终终点的关联性进行较准确的估计,避免对治疗效应点估计的影响。
猜你喜欢
故事作文·低年级(2023年1期)2023-02-23 07:18:36
基层中医药(2020年5期)2020-09-11 06:32:04
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
当代陕西(2019年14期)2019-08-26 09:41:54
阅读与作文(小学高年级版)(2019年3期)2019-04-20 13:39:30
小学生作文(低年级适用)(2017年4期)2017-07-07 10:10:41
百科知识(2015年18期)2015-09-10 07:22:44
中国合理用药探索(2012年2期)2012-03-20 16:30:30
中国合理用药探索(2011年9期)2011-03-20 16:30:26
