
Ni基非负载型催化剂合成条件对微观结构及加氢性能的影响
2018-06-09 07:35黎胜可王荧光
精细石油化工 2018年3期
石 闯,鲍 伟,施 岩*,黎胜可,孙 娜,王荧光
(1. 辽宁石油化工大学化学化工与环境学部,辽宁 抚顺 113001; 2.中石油抚顺石化分公司,辽宁 抚顺 113003; 3.中油辽河工程有限公司油气加工所,辽宁 盘锦124010)
随着世界各国对燃料中硫含量的控制越来越严格,深度脱硫已为大势所趋[1]。加氢脱硫是深度脱硫工艺中的有效技术手段。相应催化剂以是否使用载体来区分,可分为负载型催化剂和非负载型催化剂2类。目前工业上以负载型催化剂的应用居多,其合成方法通常以浸渍法为主,将活性组分负载于γ-Al2O3或Ti、Si等氧化物载体上[2],活性组分受负载量和载体相互作用的双重限制,难以满足对劣质柴油深度加氢的需求。相反,非负载型催化剂不使用载体,活性组分更为密集且不受与载体相互作用的限制,已逐渐成为加氢催化剂研究领域的热点[3]。
采用共沉淀法制备非负载型催化剂时,微观结构是影响其加氢性能的关键,较大的比表面积、孔容、孔径,均匀分散的金属活性组分,良好的活性相形貌均有利于提高催化剂的加氢性能,尤以金属摩尔配比、合成温度对催化剂的微观结构影响较为显著。在不同金属摩尔配比下,催化剂活性组分间复合程度差异较大对催化剂孔结构和比表面积产生影响。故以不同金属摩尔配比及反应温度制备Ni基非负载型催化剂,采用不同手段进行表征、分析,针对制备过程中两个关键条件对催化剂微观结构和加氢性能的影响进行系统研究。
1 实验部分
1.1 非负载型催化剂的制备
以硝酸镍、钼酸铵、偏钨酸铵为活性金属原料,采用共沉淀法制备Ni-Mo-W三金属非负载型催化剂。具体过程如下,取一定质量钼酸铵、偏钨酸铵溶于去离子水,加热搅拌至溶解,逐滴加入氨水调节pH=10,得到溶液A。另取一定质量硝酸镍溶于去离子水,搅拌至溶解得溶液B。将溶液B于同温缓慢滴入溶液A,可见有沉淀逐渐生成。连续搅拌若干小时,搅拌结束后在适宜温度下老化一定时间并在110 ℃干燥12 h[5],经焙烧后对其进行压片、过筛,经器内还原得到催化剂。将不同金属摩尔配比下的试样分别标记为Ni-Mo-W-1∶1∶1,Ni-Mo-W-2∶1∶1,Ni-Mo-W-3∶1∶1(Ni∶Mo∶W);不同合成温度下的试样分别标记为Ni-Mo-W-70,Ni-Mo-W-80,Ni-Mo-W-90(70、80、90 ℃);
1.2 催化剂的表征
采用Rigaku D/max-RB型X射线衍射仪对试样的物相结构进行表征。试样的比表面积、孔容及孔径测定在Micromeritics ASAP2405型多功能吸附仪上进行,吸附温度为-196 ℃,吸附介质为高纯氮,相对压力P/P0在0~0.995,试样的比表面积用BET法计算。采用Nicolet-58SXC型分析仪测定试样的酸类型,NH3升温吸脱附分析在Autochem2910型升温脱附分析仪上进行,升温范围为120~600 ℃,升温速率为10 ℃/min。采用日立公司S-4800型扫描电子显微镜表征催化剂的活性相形貌。
1.3 催化剂的活性评价
催化剂的加氢脱硫活性评价在实验室自建的固定床高压微型反应装置中进行,催化剂形态为片状,粒度20~40目,装填量10 ml。配制含3% CS2的环己烷溶液为预硫化液,预硫化条件为4 MPa,2 h-1,氢油比G/L=500∶1,在230 ℃下预硫化4 h,在320 ℃下预硫化6 h后泵入油品。以大连西太平洋催化剂裂化柴油为评价原料,在固定床加氢微型反应装置上对一系列加氢脱硫催化剂的脱硫活性进行评价。反应在320~380 ℃,4 MPa,1 h-1,氢油比G/L=500∶1的条件下进行。
2 结果与讨论
2.1 金属摩尔配比对催化剂微观形貌及加氢脱硫性能的影响
2.1.1催化剂的XRD表征
图1为以不同金属摩尔配比制备的Ni-Mo-W非负载型催化剂的XRD谱。由图1可见,在不同金属摩尔配比下,Ni-Mo-W三金属非负载型催化剂均在2θ=14.1°、33.9°、40.9°、60.2°等处出现不同强度的衍射峰,归属于MoS2/WS2物相,由于MoS2、WS2的衍射峰位置基本一致,在此不加以区分。另外,催化剂均在2θ=22.6°、31.5°、50.7°、56.2°处出现衍射强度稍弱的特征峰,经比对归属于Ni3S2物相。
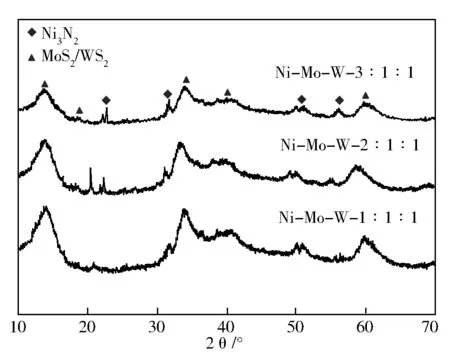
图1 不同金属摩尔配比制备Ni-Mo-W非负载型催化剂的XRD谱
在较小的金属摩尔配比下(Ni∶Mo∶W=1∶1∶1),Ni3S2对应的衍射峰强度较低,22.6°、56.2°处的衍射峰几乎无法被检测到。随金属摩尔配比的增大,Ni3S2的衍射峰强度逐渐增强,当Ni∶Mo∶W=3∶1∶1时,Ni3S2与MoS2/WS2衍射峰强度的差距最小,含量更为接近。有研究认为,Ni作为助催化组分,其硫化物Ni3S2对Mo、W活性相可起到重要的支撑、分散作用[6]。但有学者认为,Ni3S2自身仅具有很低的加氢脱硫活性,Ni-S化物的增多对提高催化剂的加氢脱硫活性十分不利[7]。另外,不同金属摩尔配比下Ni-Mo-W非负载型催化剂的衍射峰均呈低宽、致密的漫射峰形态。Amaya等认为类似衍射峰形态应归属于非晶态物质,并可用Sherrer公式(D=Kγ/Bcosθ)对其颗粒尺寸进行计算,半高峰宽B越大,对应结构的颗粒尺寸D就越小[8]。随金属摩尔配比的增大,催化剂颗粒尺寸更为精细。当Ni∶Mo∶W=2∶1∶1时,催化剂中Ni3S2物相含量适中,同时催化剂颗粒尺寸较小,对于发达孔隙结构和良好活性相形态的形成十分有利。
2.1.2催化剂的TEM表征
图2为以不同金属摩尔配比制备的非负载型催化剂的TEM。图2中均可见一定数量的黑色条纹堆叠,其条纹晶面间距均匀,为典型的MoS2/WS2活性相[9]。在较小的金属摩尔配比下,催化剂中MoS2/WS2活性相分布稀疏且堆叠层数较低,活性相晶片多呈较长的直线型排列堆积。当Ni∶Mo∶W=2∶1∶1时,活性相在TEM照片中的分布十分密集且较为均匀,堆叠层数明显增高并呈现一定曲率。随金属摩尔配比的进一步增大,催化剂活性相密集程度有所降低,堆叠层数亦有所减少。图中黑色块状区域显示其活性组分出现集聚、分布不均。在目前对Mo、W硫化态活性相的研究中,活性相条纹的堆叠层数与曲率被认为是决定催化剂加氢脱硫活性的关键[10,11]。较高的活性相堆垛数对加氢脱硫(HYD)路径脱硫十分有利,而较大的曲率则利于MoS2/WS2活性相拐点位的暴露,进而影响催化剂氢解脱硫(DDS)路径脱硫的效率。显然,Ni∶Mo∶W-2∶1∶1的活性相形态更利于其深度脱硫活性的发挥。在较小的金属摩尔配比下,活性组分无法充分复合,不利于具有良好形态MoS2/WS2活性相的形成。而在较大的金属摩尔配比下,Ni-S化物的增多使活性相分布不均,导致催化剂活性相堆叠层数下降,影响催化剂的加氢脱硫活性。

图2 不同金属摩尔配比制备Ni-Mo-W非负载型催化剂的TEM照片
2.1.3催化剂的织构性质
图3为以不同金属摩尔配比制备的非负载型催化剂的N2吸脱附等温线。由图3可见,以不同金属摩尔配比制备的三金属非负载型催化剂的N2吸脱附等温线类型均为第Ⅳ型[12],表明其均存在一定数量的介孔结构。另外,3者均在中比压附近(P/P0=0.4)出现形状相似的滞后环,其面积大小的差异表明以不同金属摩尔配比制备的非负载型催化剂在N2脱附过程中出现了不同程度的毛细凝聚。当Ni∶Mo∶W=2∶1∶1时,滞后环面积相对最大,此时毛细凝聚现象最为显著,即Ni-Mo-W-2∶1∶1具有更为发达的孔隙结构。在较小的金属摩尔配比下,催化剂在各相对压力下对N2的吸附量较小,这可归因于活性组分间未能充分反应,无法形成有效的介孔结构。当Ni∶Mo∶W=3∶1∶1时,催化剂孔隙结构的发达程度亦不理想。Yi[13]在相关研究中认为,过量的Ni使催化剂Ni-S化物含量增加,对提高催化剂的比表面积十分不利。另外,在对催化剂的孔径分布统计中发现,以不同金属摩尔配比制备的催化剂最可几孔径均为3.7 nm。故可认为该直径的介孔属于催化剂颗粒本身,其数量同样代表催化剂孔隙结构的发达程度。显然,Ni-Mo-W-2∶1∶1在该直径处的介孔数量具有明显优势。
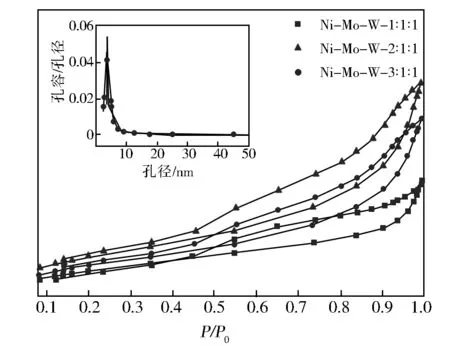
图3 不同金属摩尔配比制备Ni-Mo-W非负载型催化剂的N2吸脱附等温线
2.1.4催化剂的加氢脱硫活性评价
表1为催化裂化柴油在不同金属摩尔配比的Ni-Mo-W非负载型催化剂上加氢产物的性质。由表1可见,以Ni∶Mo∶W=2∶1∶1制备的催化剂表现出相对最佳的加氢脱硫活性,其对催化裂化柴油的最高脱硫率可达96.9%,加氢柴油密度降低了2%,同时十六烷值显著提升。在较小的金属摩尔配比下制备的催化剂对劣质柴油中有机硫化物的脱除能力有限,相应的脱硫率仅能达到94.2%,十六烷值提升幅度较小。
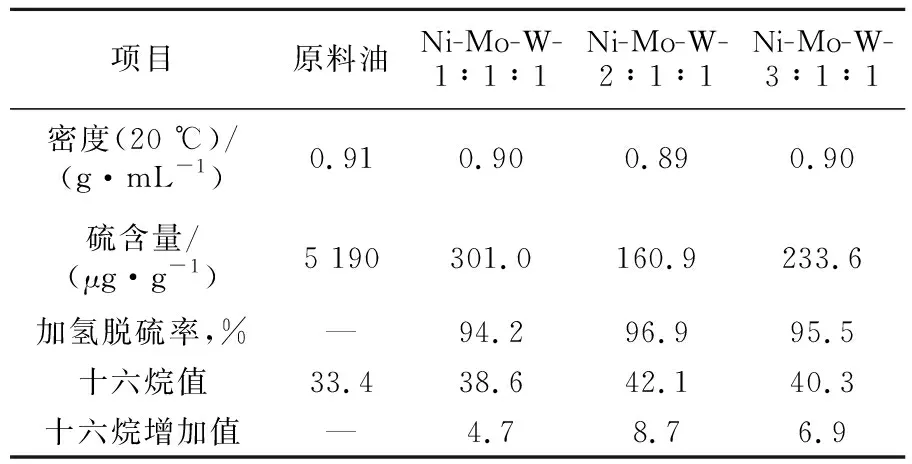
表1 催化裂化柴油在不同催化剂上加氢产物的性质
而以较大金属摩尔配比制备的催化剂加氢脱硫效果同样不够理想,加氢柴油在相同条件下的残硫量可达233.6 μg/g,柴油中的多环芳烃通过加氢饱和部分转化为饱和烃类,使十六烷值提升,但其幅度仍小于Ni∶Mo∶W-2∶1∶1。相比于Ni-Mo-W-1∶1∶1不够充分的活性组分间复合程度,Ni-Mo-W-3∶1∶1中过量的Ni使Ni-S化物增多,虽然其活性组分充分复合且具有相对发达的孔隙结构,但仍无法充分发挥非负载型催化剂的深度脱硫潜力。从脱硫活性评价结果来看,Ni∶Mo∶W-2∶1∶1具有相对最佳的加氢脱硫活性。
2.2 反应温度对催化剂微观形貌及加氢脱硫性能的影响
2.2.1催化剂的H2-TPR表征
图4为不同温度制备Ni-Mo-W非负载型催化剂的H2-TPR曲线。由图4可见,催化剂在氧化态下的TPR曲线呈双峰分布,380 ℃左右的低温还原峰是八面体配位的Mo/W还原所形成的,该配位方式的Mo/W通常被认为具有更好的供电子能力,易于硫化形成良好的微观结构,而790 ℃左右的高温还原峰表征了Mo/W四面体配位的还原,这种配位方式的Mo/W不利于形成良好的活性相形貌[14]。随反应温度的增加,催化剂的高温还原峰逐渐降低,耗氢量显著减少,但低温还原峰却有所增高,耗氢量增大,说明催化剂中八面体配位的Mo/W增多而四面体配位的Mo/W减少,对活性组分的还原十分有利。同时,随反应温度的增高,催化剂的两个还原峰逐渐向低温方向略微偏移,说明此时催化剂活性组分分布均匀,相互作用程度降低,同样利于催化剂的硫化还原和形成良好的微观形貌。故催化剂的制备宜在较高的反应温度下进行,在水浴加热的有限条件下,反应温度90 ℃时更利于活性组分间的充分复合,可形成具有多层多缺陷位的钼钨酸镍对于硫化还原十分有利。
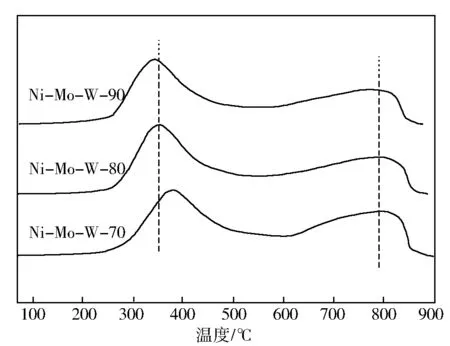
图4 不同温度制备Ni-Mo-W非负载型催化剂的H2-TPR曲线
2.2.2催化剂的XRD表征
图5为不同温度制备Ni-Mo-W非负载型催化剂的XRD谱。Ni-Mo-W三金属非负载型催化剂均在2θ=14.1°、33.9°、40.9°、60.2°等处出现不同强度的衍射峰,另外,在2θ=22.6°、31.5°、50.7°、56.2°处出现衍射强度稍弱的特征峰,分别归属于MoS2/WS2和Ni3S2物相。相比于金属摩尔配比,不同反应温度并未使催化剂的物相组成出现明显差异,但对其颗粒的精细程度影响显著。随反应温度的升高,催化剂衍射峰逐渐宽化,颗粒尺寸减小,这可归因于相应氧化态催化剂的良好还原性。精细的颗粒尺寸使催化剂活性组分分布均匀,利于发达孔隙结构的形成和酸性位的暴露,对非负载型催化剂的深度脱硫十分有利。
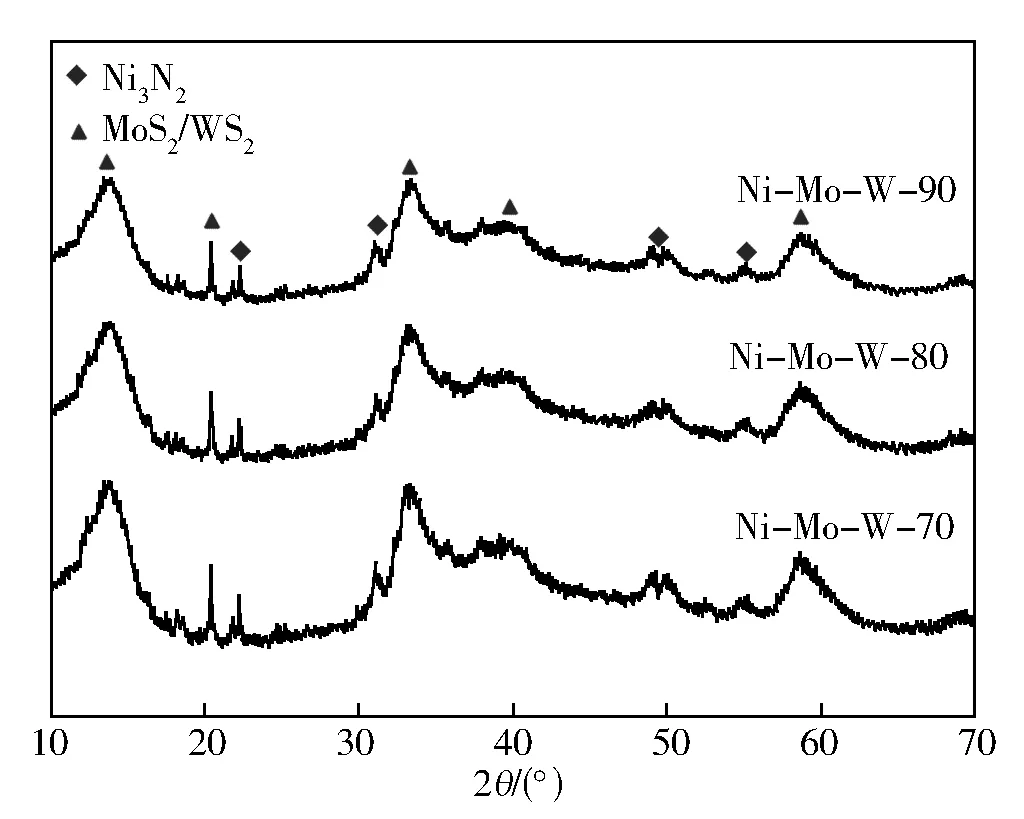
图5 不同温度制备Ni-Mo-W非负载型催化剂H2-TPR曲线
2.2.3催化剂的织构性质
表2为不同温度制备非负载型催化剂的织构性质。由表2可见,在较低的反应温度下,催化剂的孔隙结构显然不够发达,其比表面积仅为82 m2/g,孔容仅为0.11 cm3/g。随反应温度的升高,催化剂的孔隙结构显著优化。当反应温度为90 ℃时,催化剂的比表面积可达102 m2/g,孔容为0.14 cm3/g,孔径为6.5 nm,相比之下均有明显提升。催化剂的比表面积虽不是决定其加氢脱硫性能的唯一因素,但比表面积较大利于硫化物在催化剂表面的流通,较大的孔容、孔径则利于大分子硫化物的进入和吸附。故Ni-Mo-W-90的织构性质显示其具有更好的深度加氢潜力,与XRD的表征结构相一致。
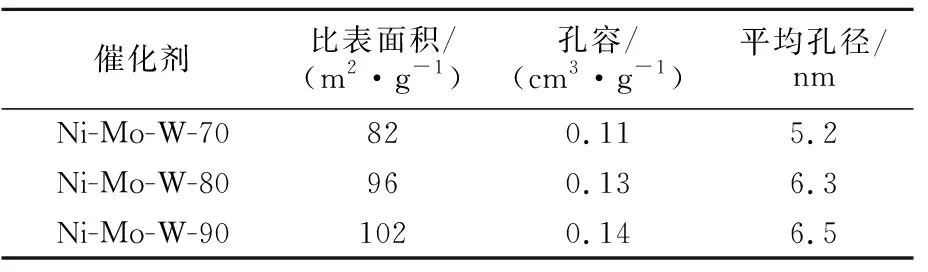
表2 不同温度制备Ni-Mo-W非负载型催化剂的织构性质
2.2.4催化剂的NH3-TPD表征
图6为不同温度制备Ni-Mo-W非负载型催化剂NH3-TPD曲线。如图6所示,以不大于80 ℃的反应温度制备非负载型催化剂的NH3-TPD曲线呈双峰分布,在175 ℃以下NH3的脱附量较多,说明其具有相对较多的弱酸酸性中心。随反应温度的升高,催化剂的NH3-TPD曲线呈多峰分布,在175 ℃以下NH3的脱附量相对较少,而在325 ℃和450 ℃左右NH3的脱附量明显增加,这说明Ni-Mo-W-90具有更多的中强酸和强酸酸性中心。故反应温度对催化剂的酸强度分布十分关键,随反应温度的提高,催化剂活性组分充分复合,分布均匀,形成了发达的孔隙结构,暴露了更多的强酸酸性中心,这对于一些复杂的大分子有机硫化物的吸附十分有利。
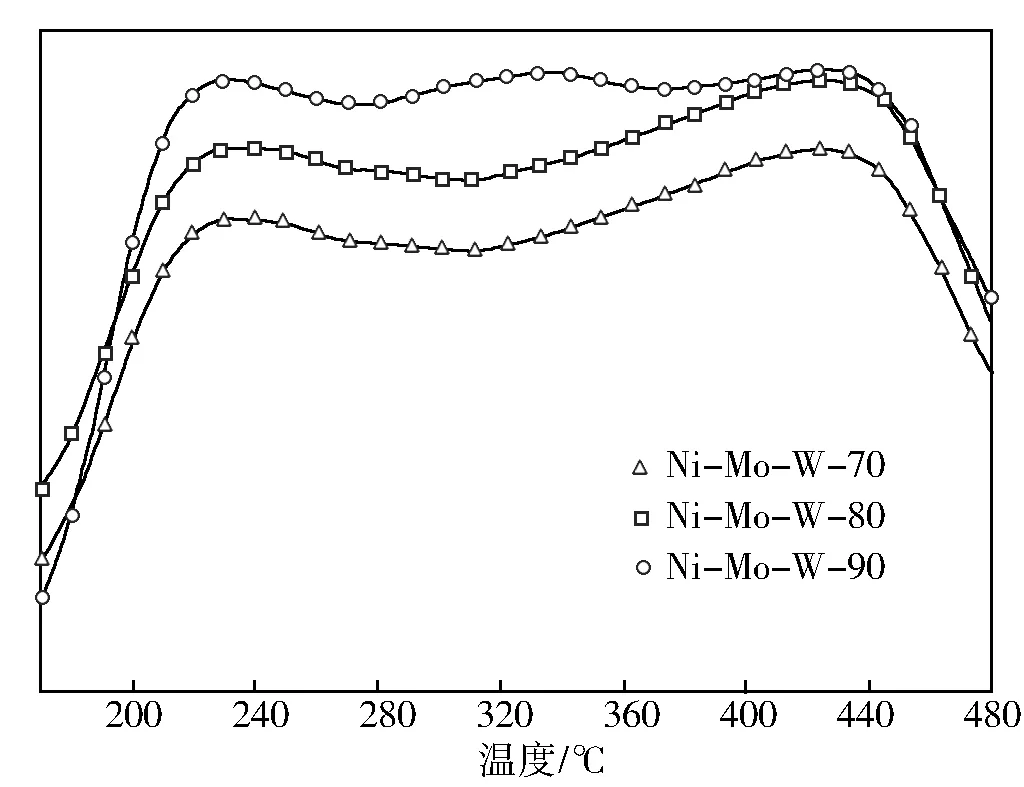
图6 不同温度制备Ni-Mo-W非负载型催化剂的NH3-TPD曲线
2.2.5催化剂的加氢脱硫活性评价
图7为不同温度制备Ni-Mo-W非负载型催化剂的脱硫率曲线。对不同催化剂的加氢脱硫性能评测均在相同工艺条件下进行,反应持续至预硫化过程结束后210 h。由图7可见,以较低反应温度制备的非负载型催化剂对劣质柴油的加氢脱硫活性并不理想,Ni-Mo-W-70和Ni-Mo-W-80随运转时间的增加活性降低明显,相应的加氢柴油中残硫量分别可达244和161 μg/g。相比之下,Ni-Mo-W-90对催化裂化柴油的加氢脱硫活性明显更优,且在考察时间内始终保持较高的反应活性,加氢柴油残硫量仅为46.7 μg/g。显然,反应温度的提升利于提高Ni-Mo-W非负载型催化剂的加氢脱硫活性。
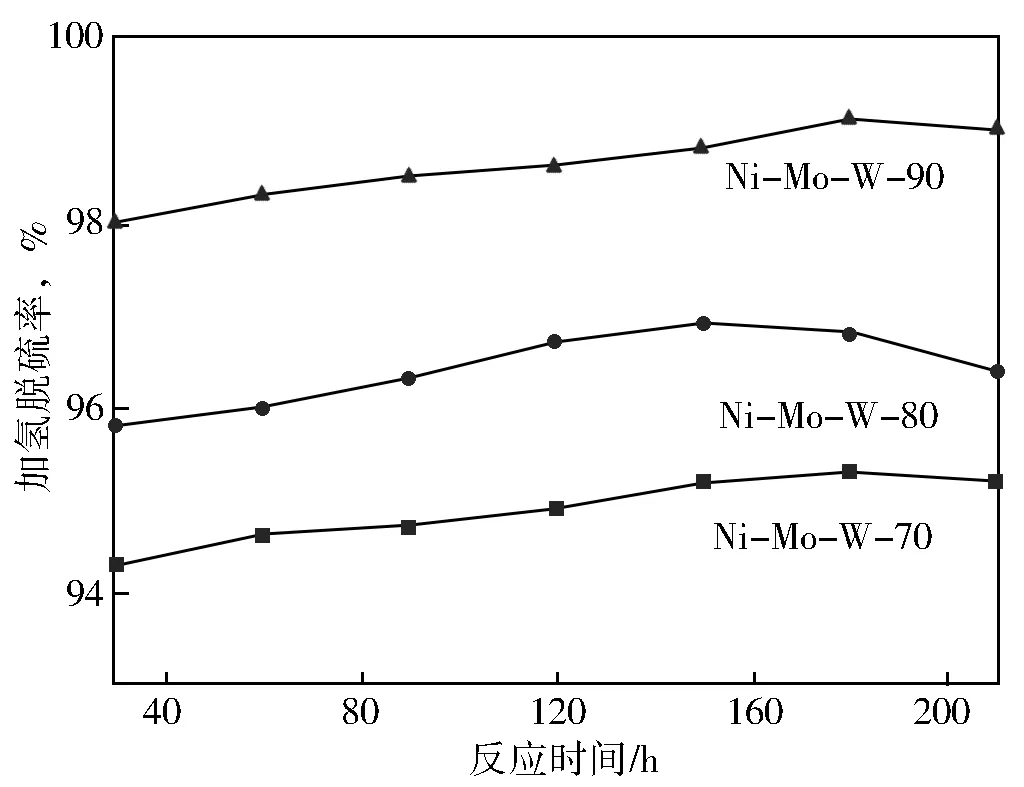
图7 不同温度制备Ni-Mo-W非负载型催化剂的脱硫率(反应条件: 4 MPa, 340 ℃, 1 h-1, G/L=500∶1)
3 结 论
采用共沉淀法以Ni∶Mo∶W=2∶1∶1制备的Ni-Mo-W非负载型催化剂中Ni-S化物含量适宜,孔隙结构发达,其MoS2/WS2活性相分布均匀,堆叠层数较高且具有一定曲率。活性评价结果表明Ni∶Mo∶W-2∶1∶1具有相对最佳的加氢脱硫活性。提高反应温度利于活性组分复合和Mo/W八面体配位形成,使催化剂硫化还原能力增强。催化剂微观结构更趋发达,中强酸和强酸酸性中心显著增加。活性评价结果显示Ni∶Mo∶W-90的加氢脱硫活性最优,对催化裂化柴油的脱硫率可达99.2%,十六烷值显著提升。
参 考 文 献
[1] 李贺, 殷长龙, 赵蕾艳, 等. 非负载型加氢精制催化剂的研究进展[J]. 石油化工, 2013, 42(7): 811-817.
[2] Chao L, Zhang Z M, Huang Y, et al. Support effects on thiophene hydrodesulfurization over Co-Mo-Ni/Al2O3and Co-Mo-Ni/TiO2-Al2O3catalysts[J]. Chinese Journal of Chemical Engineering, 2014, 22(4): 383-391.
[3] 王晨, 王海彦, 施岩, 等. PEG对Ni-Mo-W 非负载型催化剂加氢脱硫性能的改进研究[J]. 天然气化工, 2017,2 (42): 45-49.
[4] Cervantes-Gaxiola M E, Arroyo-Albiter M, Pérez-Larios A, et al. Experimental and theoretical study of NiMoW, NiMo, and NiW sulfide catalysts supported on an Al-Ti-Mg mixed oxide during the hydrodesulfurization of dibenzothiophene[J]. Fuel, 2013, 113(2): 733-743.
[7] Wang C, Wu Z, Tang C, et al. The effect of nickel content on the hydrodeoxygenation of 4-methylphenol over unsupported NiMoW sulfide catalysts[J]. Catalysis Communications, 2013, 32(2): 76-80.
[8] 王欣, 张舜光, 侯凯湖. 非负载Ni(Co)-Mo-Al2O3纳米催化剂的制备及其生物油模型化合物加氢脱氧性能研究[J]. 分子催化, 2010, 2(4): 153-157.
[9] Huirache-Acuna R, Albiter M, Ornelas C, et al. Ni(Co)-Mo-W sulphide unsupported HDS catalysts by ex situ decomposition of alkylthiomolybdotungstates[J]. Applied Catalysis A: General, 2006, 308(6): 134-142.
[10] Ledoux M J, Peter A, Blekkan E A, et al. The role of the nature and the purity of the alumina support on the hydrodesulfurization activity of CoMo sulfides[J]. Applied Catalysis A: General, 1995, 133(2): 321-333.
[11] Wang C, Wu Z, Tang C, et al. The effect of nickel content on the hydrodeoxygenation of 4-methylphenol over unsupported NiMoW sulfide catalysts[J]. Catalysis Communications, 2013, 32(2): 76-80.
[12] Sing K S. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984)[J]. Pure and Applied Chemistry, 1985, 57(4): 603-619.
[13] Yi Y, Zhang B, Jin X, et al. Unsupported Ni-Mo-W sulfide catalysts for hydrodesulfurization of dibenzothiophene by thermal decomposition of thiosalts[J]. Journal of Molecular Catalysis A: Chemical, 2011, 351(2): 120-127.
[14] Zuzaniuk V, Prins R. Synthesis and characterization of silica-supported transition-metal phosphides as HDN catalysts[J]. Journal of Catalysis, 2003, 219(1): 85-96.
猜你喜欢
河北画报(2020年10期)2020-11-26
中国特种设备安全(2019年3期)2019-04-22
石油知识(2019年1期)2019-02-26
橡塑技术与装备(2018年1期)2018-12-25
英美文学研究论丛(2018年2期)2018-08-27
山东工业技术(2016年15期)2016-12-01
通信电源技术(2016年4期)2016-04-04
智能建筑电气技术(2015年5期)2015-12-10
橡胶科技(2015年6期)2015-07-31
雕塑(2000年4期)2000-06-24
