
乌桕籽油制备环氧脂肪酸乙酯及增塑PVC研究
2018-06-09 07:35:14黄金瑞聂小安蒋剑春杨天生
精细石油化工 2018年3期
李 科,陈 洁,黄金瑞,聂小安,2*,蒋剑春,2,杨天生
(1.中国林业科学研究院林产化学工业研究所,江苏 南京 210042;2. 中国林业科学研究院 林业新技术研究所,北京 100091;3.南京林业大学化学工程学院,江苏 南京210037)
环氧脂肪酸甲酯[1]( EFAME)等环氧增塑剂产品已经逐步成熟并运用于PVC加工中,且大多数含不饱和双键的油脂都可以用作制备原料[2-4]。这其中就包括餐饮废弃油脂和木本油脂[4],其工艺也几乎相似,不必过多研究。虽然对比邻苯类增塑剂来说较为健康环保,但人们忽视了原料甲醇的毒性及甲酯的降解产物也可能含有毒性的甲醇。相对于甲酯,环氧脂肪酸乙酯(EFAEE)具有更好的安全性,且鲜见相关报道。在众多的木本油脂中,乌桕籽油的脂肪酸成分与众不同[5],这主要是因为梓油中含有约30%的甘油四酯(见图1),其脂肪酸组成中含有低碳数的2,4-癸二烯酸和8-羟基-5,6-辛二烯酸,其对应的甲(乙)酯等沸点极低,从而可能影响到相应增塑剂闪点等性能。为探究这一问题,本文采取了分离2,4-癸二烯酸乙酯的方法,分离出的2,4-癸二烯酸乙酯可作为增香剂原料来使用,可降低生产成本,提高乌桕籽油的使用价值。

图1 乌桕籽油中四酯的结构示意
1 实 验
1.1 试剂与仪器
乌桕籽油(酸值(KOH)为12.8 mg/g,碘值为181),武汉远成共创科技有限公司;无水乙醇、甲醇钠、对甲苯磺酸、甲酸、双氧水、碳酸钠等均为分析纯。
DF-101S集成式恒温加热磁力搅拌器,巩义市英峪予华仪器厂;Agilent 6890/5973型气相-质谱联用仪,美国安捷伦;3400-Ⅰ型扫描电镜,日本日立公司;D8 FOCUS型X射线衍射仪等,德国Bruker公司。
1.2 实验步骤
1.2.1预处理
由于原料油脂的初始酸值偏高,在酯交换前需要进行酯化等预处理。具体方法为向装有温度计、冷凝管和搅拌器的250 mL三口烧瓶中加入梓油、乙醇和催化剂对甲苯磺酸,搅拌加热至70 ℃反应至酸值(KOH)小于1 mg/g后降至室温置入分液漏斗,水洗两次取上层减压蒸馏脱水,其目的在于减少产品中的皂含量,便于后续酯交换甘油的分离。
1.2.2梓油乙酯的制备
在装有温度计、冷凝管和搅拌器的250 mL三口烧瓶中,加入一定质量降酸预处理后的乌桕梓油、乙醇和催化剂甲醇钠(n(梓油)∶n(乙醇)=7∶1,催化剂用量为梓油质量的1.6%),水浴加热,在65 ℃反应2 h,减压脱除乙醇,转化率[6]为76.7%。将混合物置入分液漏斗加水分层,取上层并真空脱除水和2,4-葵二烯酸乙酯等再减压蒸馏至250 ℃既得梓油乙酯,。
1.2.3环氧梓油乙酯的制备
向250 mL 三口烧瓶中加入上述梓油乙酯50 g,适量的甲酸和催化剂对甲苯磺酸,升温至50 ℃,滴加计量的双氧水;滴完后升温至反应设定的温度,到预定时间即可停止;倒出物料静置分层,用5%碱液碱洗上层油相至pH 值到6.5左右;再分离下层水相,水洗上层至中性;然后在90 ℃下减压蒸馏至无水,剩余淡黄色透明油状液体即为环氧脂肪酸乙酯。
1.2.4样条制备及性能测定
软质PVC薄膜的配方参见表1(E表示乙酯,数字表示环氧乙酯占增塑剂百分比),在高速混合机中搅拌混匀2 min后,在利用双辊开炼机在165 ℃下混炼3~8 min并制成有一定厚度(约0.2 mm)的PVC薄膜,再通过模具将PVC薄膜切成哑铃型试样,进行力学性能测试。
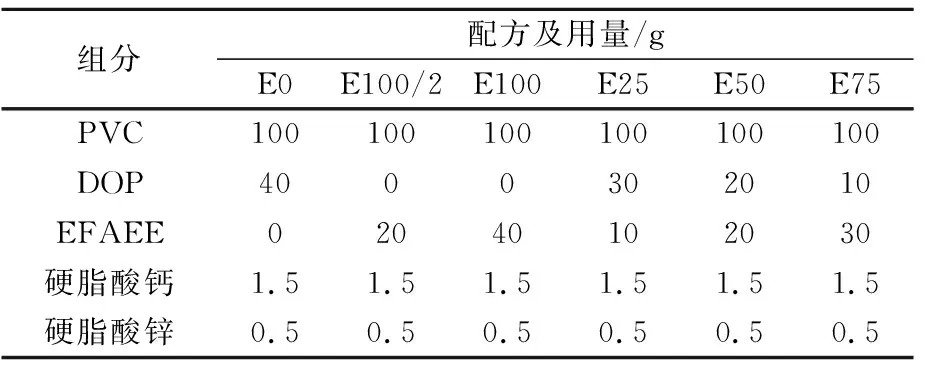
表1 软质PVC膜配方
在试验中涉及对酸值、环氧值、碘值等指标的检测,测定方法参照GB/T 1668—95增塑剂酸值及酸度的测定,GB/T 1677—2008(盐酸-丙酮法)增塑剂环氧值的测定,GB/T 5532—2008动植物油脂碘值测定法等标准进行;
采用扫描电镜(SEM)观察增塑后PVC断裂面形貌;
采用X射线衍射技术表征纯PVC及增塑PVC的物相结构,扫描速度为6(°)/min;
采用薄膜拉伸模式在DMA-Q800 动态力学分析仪上进行动态力学性能(DMA)测试,测试频率为1 Hz,升温速率为3 ℃/min,测试温度范围为-60~80 ℃ ,每个试样(50 mm×6.2 mm×0.2 mm)进行重复测试;
热重分析:采用NETZSCH公司生产的TG209F1热重分析仪及409PC热重分析和IS 10型红外光谱仪联用仪,温度范围均为35~500 ℃,升温速率均为10 ℃/min,氮气氛。
2 结果与讨论
2.1 原料及配比分析
由于甲酯化是测定脂肪酸组成的惯用方法,所以根据文献[5]的方法将乌桕籽油合成乌桕籽油脂肪酸甲酯并进行气质分析,结合GC/MS总离子流色谱图和相应的质谱图可以通过面积归一化法都知脂肪酸甲酯的组分和对应的含量(质量分数,下同),成分及含量见表2。

表2 梓油甲酯成分及含量
注:C10∶2表示脂肪酸含10个碳,2个双键。
从表2可以看出乌桕梓油中不饱和脂肪酸占82.334%,其中多不饱和脂肪酸占53.293%,共轭组分占6.055%,是用来合成环氧脂肪酸酯的优良原料。根据各组分的相对分子质量和GC含量(其他按296计算),以及8-羟基-5,6-辛二烯酸二酯的相对分子质量(320),可计算出乌桕梓油的相对分子质量Mr约为878.48(Mr=∑MnCn×3-4,Mn为第n种脂肪酸甲酯的相对分子质量;Cn为第n种脂肪酸甲酯在总甲酯中的GC含量)。
此外,从表2还可以看出,梓油中的2,4-葵二烯酸含量约占总脂肪酸的5.208%,而未见8-羟基-5,6-辛二烯酸的踪迹,这主要是因为其在检测过程中被分解或聚合所致,但其含量与2,4-癸二烯酸相当,且同属低沸点脂肪酸,这在理论上可能影响到增塑剂产品的闪点,故在环氧化前将其进一步脱除。
2.2 梓油乙酯环氧化工艺
以甲酸用量/g(A)、双氧水用量/g(B)及反应温度/℃(C)、反应时间/min(D)为考察因素,以环氧值作为指标,设计了L9(34)正交实验(其中梓油乙酯的质量按100 g计),因素水平见表1,正交实验设计及数据处理结果见表4。

表3 正交实验因素水平表

表4 正交实验设计及数据处理结果
从表4可以看出,影响脂肪酸辛酯环氧化效果的主次因素排序为B—A—D—C,最优化工艺为A2B3C1D1,即脂肪酸乙酯100 g、甲酸14 g、双氧水113 g、反应温度55 ℃,反应时间为3 h。以此条件做一组实验,结果环氧值为5.5,由于实验波动和误差略低于实验5、6,但在表4中已处于较高水平,这说明所得工艺适合合成环氧脂肪酸乙酯。
2.3 结构表征
在制备脂肪酸乙酯脱除甘油时采取了加水促进分离,为了确认乙酯的生成及转化情况,对酯交换前后的梓油(三酯)及产物乙酯(单酯)进行了凝胶色谱分析,结果见图2。

图2 梓油及其酯交换产物的凝胶色谱
从图2可以看出,反应前的梓油相对分子质量较高,因为其主要成分为甘油三酯结构;而经过与乙醇的酯交换后,其产物相对分子质量仅为335,降低了近2/3,与结构变化完全一致,这充分说明了此时的产物大都为单酯结构的脂肪酸乙酯。
此外,环氧化效果较难判定,为确定产物环化效果,对乌桕梓油脂肪酸乙酯及环氧化后的产物做了红外分析对比,测试波段为500~3 500 cm-1,其结果见图3。

图3 环氧化前后红外光谱
由图3可以看出,脂肪酸乙酯在3 009 cm-1处的C—H键的伸缩振动峰在环氧乙酯中已经消失;且环氧脂肪酸乙酯在指纹区719 cm-1处的双键伸缩振动峰也明显弱于脂肪酸乙酯;此外,环氧脂肪酸乙酯在822 cm-1处出现了环氧基团的特征吸收峰,且在环氧乙酯中未见开环所形成的醇羟基特征峰。
2.4 PVC制品分析
2.4.1结晶状态分析
为确定常用增塑剂DOP及所合成的环氧脂肪酸乙酯对PVC结构的影响,对其增塑的PVC薄膜及未增塑的PVC进行了X射线衍射扫描,结果参见图4。

图4 PVC及增塑PVC薄膜的 X射线衍射谱
从图4可以看出,所有PVC试样的出峰都集中于小衍射角,且均在2θ=24.1°附近出现了典型的散射驼峰,表明PVC材料基本上是无定型聚合物;但纯PVC在2θ=17.1°和19.1°附近出现了小而弱的结晶衍射峰,17.1°处峰高略大于19.1°处,这是PVC链段的结晶峰,且相互之间有一定的叠加,这说明在纯的PVC中有少部分结晶,这是由于分子内、分子间的氢键相互作用而使结晶得以稳定存在的结果;对比所有增塑PVC的XRD谱图,可以看出增塑剂对PVC薄膜聚集态的影响不明显;但也可发现环乙酯(EFAEE)与DOP增塑薄膜的谱图更为接近,及DOP和环乙酯增塑的PVC在17.1°处的衍射峰均明显减弱(或消失),这说明这环乙酯与DOP一样明显降低了PVC材料的结晶度,应该具有良好的增塑性。
2.4.2拉伸试验及断面分析
所得环氧脂肪酸乙酯为黄色透明液体,环氧值约在5.5。X射线衍射测试已经足以表明其与DOP一样可明显改变PVC塑料的结晶性。为进一步确定所合成的环氧脂肪酸乙酯的运用性能,对环氧脂肪酸乙酯、DOP及其复配增塑产品进行了共混压膜实验及拉伸测试,实验表明均具有良好的相容性及成膜性能,其中PVC薄膜拉伸测试结果见表5。

表5 力学实验结果
从表5可以看出,在力学性能上,单独使用EFAEE(E100)的最大力与DOP(E0)相当,拉伸强度较DOP增塑薄膜明显下降,且EFAEE增塑的PVC薄膜的断裂伸长率明显较DOP的提高了12%,这说明EFAEE具有较好的增塑性能;对比E100/2与E100可以发现,E100/2样条的厚度、最大力、强度均大于E100,而断裂伸长率又过低,这主要是因为添加量过小致使混合不均未起到良好增塑效果所致;从EFAEE与DOP复配效果可以看出,复配配方的薄膜厚度基本与E0相当,随着EFAEE的增加最大力与拉伸强度均略有下降,但断裂伸长率却明显逐步提升,这说明PVC薄膜的塑性随着EFAEE的增加而提高,且当EFAEE复配量达到50%时其PVC薄膜的断裂伸长率就已经超过了纯DOP增塑制品。综上所述,说明环氧脂肪酸乙酯(EFAEE)具有较好的增塑性能,且可根据需要与DOP复配使用。
为更好的解释EFAEE 增塑效果,对EFAEE及DOP增塑的PVC拉伸薄膜断面形貌进行了扫描电镜(SEM)拍照(1 000倍),见图5。

图5 PVC样片拉伸断面SEM照片
从图5可以看出,无论是DOP还是环氧脂肪酸乙酯增塑的PVC断口均未出现明显的断口,且具有明显的小凸起,且EFAEE增塑PVC的断面与DOP增塑的断面及其相似,这说明EFAEE与DOP一样均能够很好的溶入PVC体系并破坏其结晶性,这与XRD测试结果一致,说明EFAEE起到与DOP相似的增塑作用;此外可以看出EFAEE制品的凸起与孔径均略大且更具层次感,这说明EFAEE增塑制品应具有更好的柔韧性,与拉伸测试中断裂伸长率等结论完全相符。
2.4.3薄膜DMA测试
为更好的了解环氧脂肪酸乙酯及复配产品对PVC材料的影响,对E100/2、E100、E25、E50、E75及DOP(E0)增塑PVC薄膜进行了动态热机械分析(DMA)测试,有关其动态储能模量(E′) 、损耗模量(E″)及损耗角正切(tanδ)变化见图6。
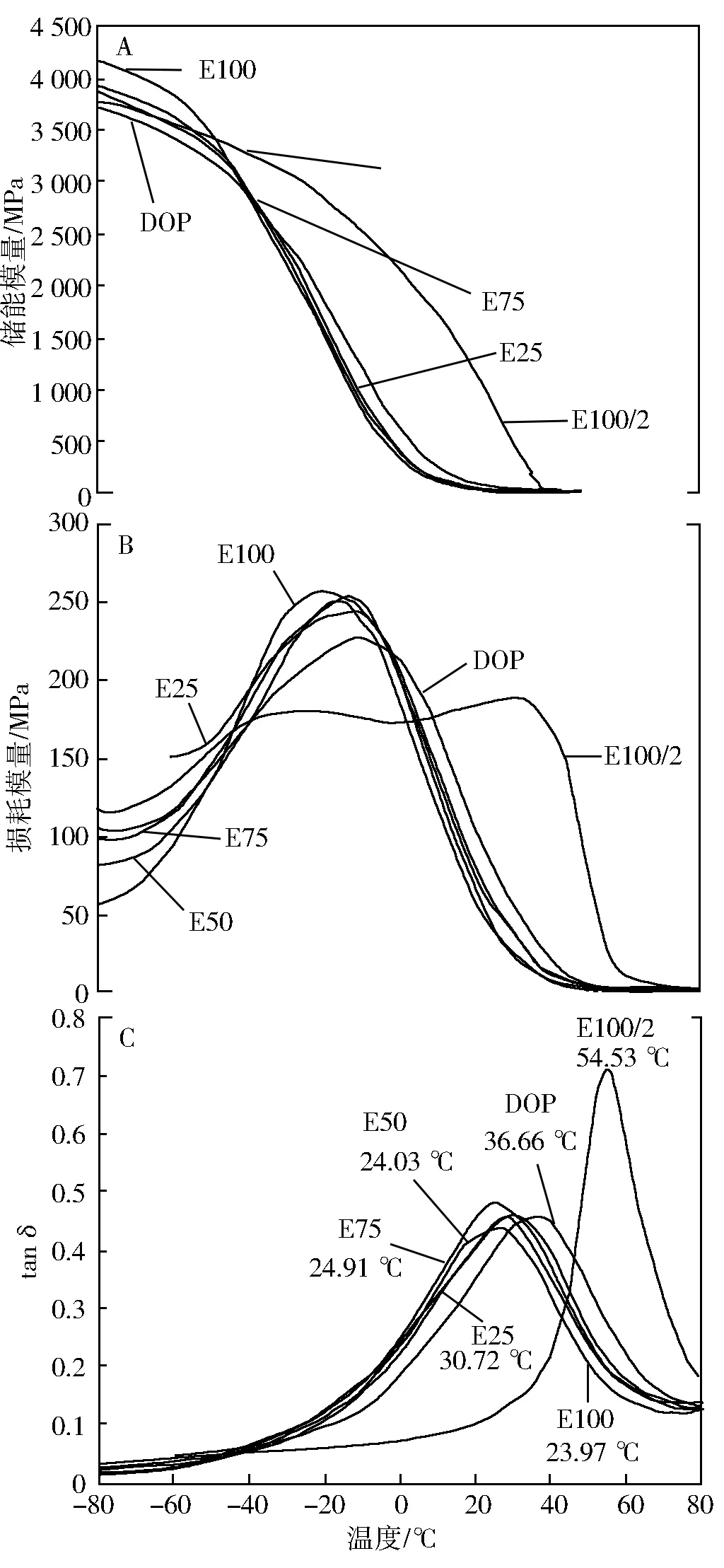
图6 PVC薄膜拉伸粘弹谱
从图6可以看出,无论是储能模量、损耗模量还是tanδ所对应的变化趋势及温度,都表明环氧脂肪酸乙酯用量在PVC质量的20%(E100/2)时增塑性能较差;图6B中高弹态和粘流态的转变温度(Tf)平台化表明增塑剂用量过少会导致材料不完全塑化,从而影响材料断裂伸长率,这与力学测试结果完全吻合;而用量在40%(E100)时环氧乙酯表现出较好的增塑性能,C图中可以看出其增塑PVC薄膜的玻璃化温度Tg较DOP的低了12.67 ℃;此外从复配的情况看,PVC薄膜总体上随着环氧乙酯对DOP比值的增加而具有更好的加工可塑性,材料的Tg也随着该比例的增加而降低。这些都充分说明了EFAEE具有较好的增塑性能,且可与DOP复配使用。
2.4.4PVC制品热稳定性
图7是DOP、EFAEE及其复配产品增塑的PVC薄膜的热重分析曲线(其中A为TG图;B为DTG图)。
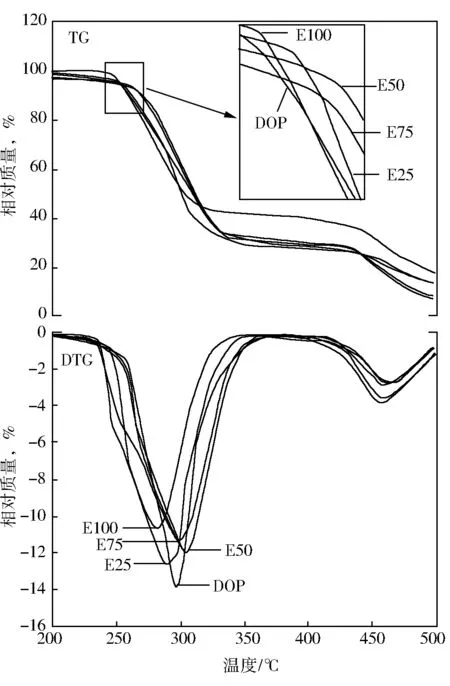
图7 PVC薄膜热重分析曲线
从图7可以看出,PVC薄膜热分解一般经历三个阶段,第一个失重台阶(240~320 ℃)主要是HCl及部分氯代烃的脱除;第二阶段为一平台(320~450 ℃),此时失重率较少,主要为第一阶段所生成的共轭多烯烃的环化、苯环化及聚合等重构反应;第三个台阶主要是PVC断链及第二阶段生成的稠环芳烃的裂解失重;此外从图7A中可以看出,单纯EFAEE增塑的PVC薄膜的第二平台相对质量明显高于所有增塑产品,这说明EFAEE具有优良的热稳定性;DOP及其与EFAEE复配产品增塑的PVC薄膜的热稳定性非常相似,这说明单纯DOP及其复配增塑剂所增塑的PVC产品具有较好的热稳定性;从图7B中可以看出,在300 ℃处,DOP增塑材料的失重率较所有含EFAEE的PVC要高,且在共混体系中失重速率随着EFAEE的增加而降低,此外从图7A中的放大图中也能看出DOP增塑材料的失重曲线更接近低温区,这说明EFAEE较DOP可以更有效的提高PVC材料的热稳定性。这主要是因为EFAEE结构中含有可束缚裂解产生的HCl的环氧环所导致的。
2.4.5PVC热分解分析
为进一步分析确认PVC薄膜热分解的历程及EFAEE增塑的PVC薄膜与DOP增塑的PVC薄膜在热解过程中的差异及产生的原因。本文采用了热失重-红外、热失重-质谱联用仪对各自热解及其产物红外质谱特征进行了测定,图谱参见图8和图9。
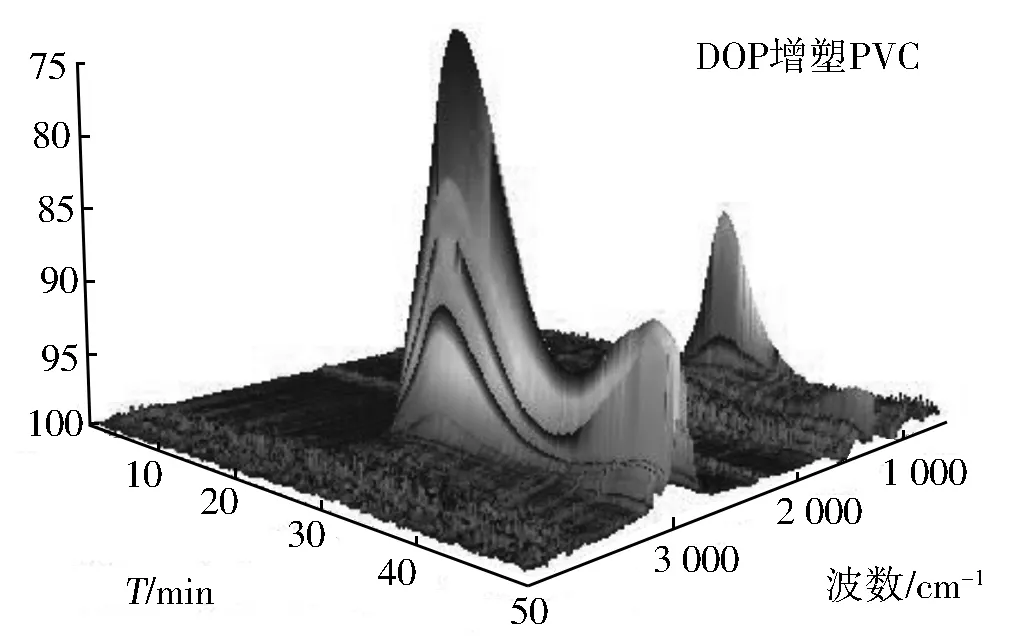
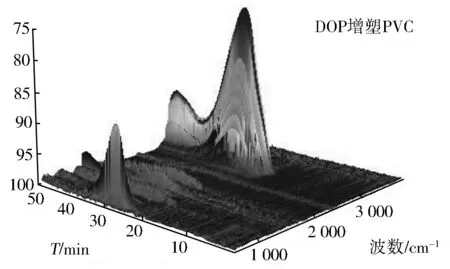

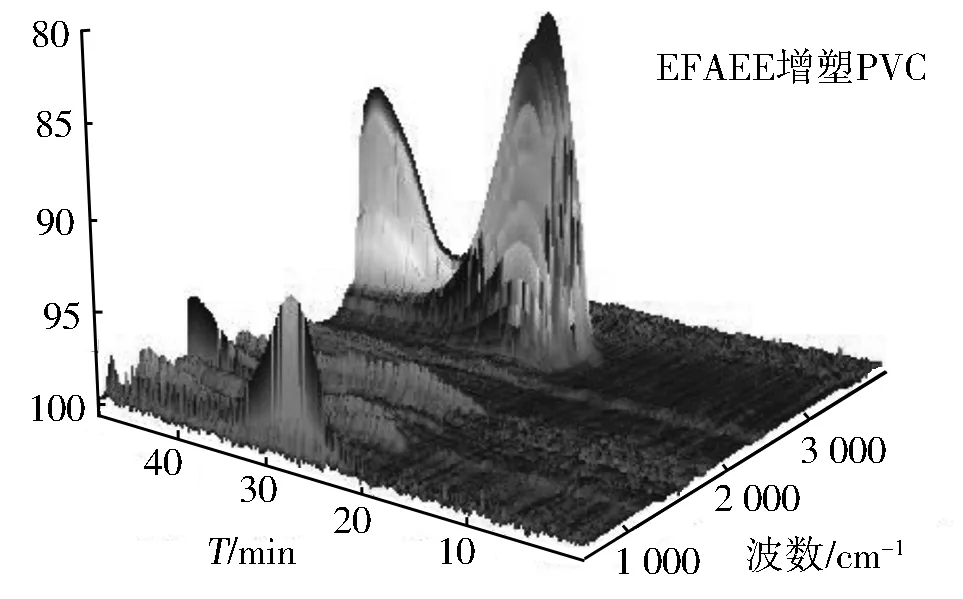
图8 DOP(上)与EFAEE(下)增塑PVC的3D TGA-FT-IR谱
图8上为DOP的TGA-FT-IR图谱,下为EFAEE的TGA-FT-IR图谱,其中左右两图分别是高波数和低波数方向视角。从图8可以看出,PVC整个热解过程中红外检测到的主要气体成分有二氧化碳(2 365,669 cm-1)、一氧化碳(2 170 cm-1)、氯化氢(2 948 cm-1)、酯类(1 780,1 290,1 150 cm-1)、苯类(3 078,1 638 cm-1)等。这是因为增塑剂未改变PVC分子结构且DOP和EFAEE均为酯类化合物;其次,可以看出PVC的裂解一旦开始会经历两个峰(台阶)和一个谷(平台)三个阶段,这与热重分析TG结果一致;此外,图9表明DOP增塑的PVC前期裂解产物较多(如300 ℃左右产生的HCl及氯代烃多于EFAEE增塑的PVC),而EFAEE增塑的PVC在高温段裂解量明显高于DOP增塑的PVC(450 ℃左右EFAEE试样中的CO2、CO及苯类明显高于DOP增塑的PVC),造成这一现象的主要原因是EFAEE增强了PVC的稳定性,使其需要在较高温度下裂解,与热重分析DTG结果完全一致。
由图9可见,在PVC热解过程中产生了水、一氧化碳、二氧化碳、氯化氢、苯等离子流,与TGA-FT-IR基本一致;此外较多的碎片主要有甲基、甲氧基、氯甲基、氯乙基、甲苯等。由PVC热解的总离子流图(TIC)及各主要离子流、碎片的色谱图可见, 其变化趋势主要有H2O、CO、HCl、CO2、氯甲基几种类型,其他甲基、甲氧基趋势类似CO,苯、甲苯、氯乙基与氯甲基类似。从TIC中可以看出,在低温阶段(1 500 s以内)EFAEE增塑的PVC热解量大于DOP,而在高温区却明显低于DOP产品,这主要是因为EFAEE易在低温区热解挥发而其中的环氧基能提高PVC的热稳定性致使其在高温阶段更加稳定;EFAEE在m/s=18处的值几乎均高于DOP,这可能是EFAEE的热解产生的水所致,而水可以有效吸收稀释自由基使体系更稳定;CO的离子流图与TIC几乎一致,且其离子流与TIC在同一量级,说明PVC热解产物以CO为主;HCl的离子流图表明EFAEE并没有有效减少HCl的排放量,这可能是因为在TGA中气体一旦产生就难以与环氧基接触所致,而CH4Cl等的离子流图则说明EFAEE可有效降低PVC中氯代烃的热分解量;此外,CO2的离子流图表明EFAEE可有效降低PVC链段的低温热解而使得其在高温阶段裂解,这与热红外结果一致。

图9 DOP与EFAEE增塑PVC的TGA-MS谱
2.4.6PVC热解活化能对比
热解活化能是衡量材料耐高温性能的一项重要参考值,活化能越高说明材料越耐高温降解。所以通过计算EFAEE和DOP增塑材料在同升温速率情况下的热解动力学参数活化能,可有效的看出两者的热稳定性差异。首先假设PVC在各阶段的热降解为一级反应,则反应速率方程为:
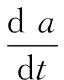
(1)
由于热分解分数a是温度T的函数,而程序升温中温度T又是时间t的函数,即a=F[f(t)]或da/dt=(da/dT)(dT/dt) =(da/dT)β(β为恒定的升温速率)。所以(1)式可表示为:
(2)
(3)

图10 PVC热解过程中ln[-ln(1-a)/T2]与1/T关系
从图10可以看出:1)PVC热解的三个阶段的活化能是完全不同的,即不同阶段应该对应着不同的反应机理;2)热解平衡阶段(第二阶段)的斜率为正值,说明该阶段发生了共轭多烯烃的环化、苯环化及聚合等重构反应;3)第三阶段的活化能很小,这看似于高温区不符,其实不然,造成这一表象的主要原因是该阶段的剩余质量较小且部分多环芳烃在继续缩合最终形成残余,大量掩盖了裂解活化能,致使总体活化能不高。所以对比热稳定性主要对比第一阶段(图10中最右侧线段)的活化能即可,从拟合方程的斜率可以计算(R=8.314 J/(mol·K))出DOP(上线)与EFAEE(下线)增塑的PVC薄膜在第一阶段的热解活化能依次是109.9 kJ/mol和125.7 kJ/mol。这充分说明了EFAEE有效的提高了PVC材料的热稳定性。
3 结 论
通过酯交换和环氧化两步反应可合成环氧脂肪酸乙酯类增塑剂,环氧化最佳工艺条件为:每百克脂肪酸乙酯需甲酸14 g,双氧水113 g,反应温度控制在55 ℃,反应时长为3 h,所得产品环氧值约为5.5;此外,乙酯中所蒸馏出的轻组分可作为原料提取增香剂2,4-癸二烯酸乙酯,用以降低生产成本,提高乌桕籽油使用价值;在增塑PVC的应用实验中,X射线衍射表明环乙酯可有效降低PVC塑料的结晶度;薄膜拉伸实验表明环氧脂肪酸乙酯(EFAEE)具有较好的增塑性能,断裂伸长率优于DOP;DMA测试也表明其增塑的PVC薄膜的玻璃化温度也明显低于DOP增塑薄膜;热重等分析及热解活化能表明EFAEE可有效提升PVC材料的热稳定性;且所有分析均表明EFAEE可根据需要与DOP复配使用。而在耐溶剂及生理毒性上尚需要进一步研究。
参 考 文 献
[1] 李祥庆, 沈凯华, 严秋钫. 环氧脂肪酸甲酯的合成研究进展及应用[J]. 塑料助剂, 2016(4):23-27.
[2] 程正载, 林素素, 王洋,等. 菜籽油非均相催化环氧化合成环氧脂肪酸甲酯[J]. 中国油脂, 2012, 37(8):49-52.
[3] 刘振兴, 聂小安, 王义刚. 白花树果油环氧脂肪酸甲酯增塑剂的制备及其应用[J]. 现代化工, 2014, 34(2):97-100.
[4] 贾茂林, 王芳, 陈意,等. 地沟油改性制备PVC用环氧脂肪酸甲酯增塑剂[J]. 中国塑料, 2014, 28(11):82-87.
[5] 尹立军, 陆向红, 卢美贞,等. 乌桕梓油中四酯的分离及鉴定[J]. 中国粮油学报, 2012, 27(8):52-56.
[6] 李科,李翔宇,蒋剑春,等.质量法:一种计算油脂甲酯化转化率的新方法研究[J].林产化学与工业,2011,31(2):43-47.
猜你喜欢
林业与生态(2022年5期)2022-05-23 01:16:51
花卉·上半月(2022年9期)2022-04-29 00:44:03
现代园艺(2017年21期)2018-01-03 06:42:18
宿州学院学报(2017年10期)2017-11-23 08:10:01
浙江农业科学(2017年5期)2017-06-21 15:12:46
中国塑料(2016年2期)2016-06-15 20:29:57
中国塑料(2015年9期)2015-10-14 01:12:31
中国塑料(2015年5期)2015-10-14 00:59:52
中国塑料(2015年5期)2015-10-14 00:59:41
中国塑料(2014年11期)2014-10-17 03:07:50
