
苯为前驱物形成二噁英的反应机理
2018-01-23 08:57高正阳韩文涛李明晖华北电力大学能源动力与机械工程学院河北保定071003
中国环境科学 2018年1期
高正阳,韩文涛,丁 艺,孙 尧,李明晖 (华北电力大学能源动力与机械工程学院,河北 保定 071003)
氯代二噁英(PCDD/Fs)一直深受人们关注,垃圾焚烧是生成二噁英类物质的主要途径,在垃圾燃烧以及热解过程中PCDD/Fs形成主要经由两种方式[1]:一种为denovo合成反应,该反应发生的条件为存在无定型碳或者石墨退化层,必须有氧源以及氯源,需要 CuCl2或者过渡金属物质的催化,反应主要发生在 200~400℃[2],这是一种高温下的非均相反应.另一种为氯化前驱物均相气态合成,其化学反应温度范围主要处于 400~800℃之间,后者的反应生成速率远远大于前者[3-4].形成二噁英的前驱物种类多样,包括脂肪族化合物、苯、带官能团的单环芳香族化合物、氯代的芳香族化合物以及蒽醌的衍生物等[5].
在前驱物合成PCDD/Fs机理研究中,氯酚为最直接的典型前驱物,因此相关研究主要以氯酚为主,其次为对氯苯的研究,对于其他前驱物机理反应报道较少.这其中 Ghorishi等[6]与 Stieglitz等
[7]分别研究了氯代芳香烃类物质1,2-二氯苯、2,4-二氯酚以及 1,2,4,5-四氯苯生成二噁英的化学反应特性,发现氯酚的催化反应活性远高于氯苯,前者二噁英生成量远大于后者;相同条件下,氯苯类前驱物生成PCDDs的反应速率比PCDFs高约 2个数量级,且大部分的二噁英均以气态形式存在. Ryan等[8]与Schoonenboom等[1,9]探索以甲苯与苯为代表的非氯代芳香烃在飞灰、CuCl2/Al2O3催化条件下的反应产物,均检测到了氯代二噁英以及氯代呋喃的存在.由于二噁英产生过程的复杂性,实验方法很难检测到反应过程中的中间产物及过渡态等物质,而量子化学克服了实验上的缺陷,从本质上揭示化学反应机理,因此很多学者采用量化计算的研究手段对不同类型二噁英前驱物的反应机理进行探究[10-12].
但是目前为止还鲜见以苯为前驱物形成PCDD/Fs的相关理论研究报道,为此本文应用量子化学方法研究了苯为前驱物二噁英气相反应机理,研究其氯化、氧化反应,生成2,3-二氯苯酚及 3,4-二氯苯酚过程,进一步以二氯苯酚为前驱物交叉反应生成PCDD/Fs的气相反应机理,并对反应涉及的基元过程做出相关动力学分析.
1 研究方法
基于前人实验结果推测PCDD/Fs很有可能经由两步反应机制,第1步是苯在催化作用下发生亲电芳香取代过程,第2步是氯化碳基的氧化分解,并进一步环化生成最终产物.采用密度泛函理论[13-14]在 B3LYP[15]/6-311+G(d,p)水平下计算获得反应路径中涉及到的反应物、过渡态、中间体、产物等各个驻点的空间结构参数.对涉及到的所有驻点结构进行了频率分析,确保反应物、中间体、产物不出现虚频,保证结构的稳定性;过渡态有且仅有一个虚频,保证过渡态结构的唯一性.过渡态是在初猜结构的基础上采用 TS算法定位并优化得到,对每个过渡态在同一方法基组水平下做内禀反应坐标(IRC)计算,验证该过渡态结构与该基元反应的反应物与产物相关联.
在 B3LYP[15]/6-311+G(d,p)水平下对各驻点结构进行了自由能以及热化学焓的计算,并考虑了零点能的校正,进一步计算得到各基元反应的反应势垒及反应热.变分过渡态理论 VTST在温度越高、势垒越低的情况下优势明显[16],本文计算采用变分过渡态理论对于关键基元反应进行化学反应速率常数的计算,并考虑隧道效应,对计算结果进行了校正,在此基础上拟合得到阿伦尼乌兹(Arrhenius)方程.计算方法与基组的稳定性、可靠性已经得到 Dar等[17]在三氯硫酚生成硫代二噁英的研究中验证.所有计算均应用Gaussian09软件包[18]在型号为 ServMaxPSC-201GAMAX服务器完成.
2 结果与讨论
2.1 苯的氯化、氧化及生成苯氧自由基
2.1.1 苯的氯化、氧化途径 在当前工业苯酚的制备过程中,采用O2、H2O2等氧化剂可以将苯直接氧化成苯酚[19-20].主要的机理为:H2O2在催化剂作用下形成的 HO·会进攻苯环,通过形成中间体羟基环己二烯自由基进而形成苯酚[21].O2的催化反应路径主要有2条,路径1为O2在催化剂作用下发生氧化还原反应生成 H2O2,进一步以H2O2为氧化剂催化制取苯酚;路径2为O2直接在催化剂条件下与苯反应而不经过形成中间反应物H2O2形成苯酚[21].
基于苯酚的制取,推测在复杂的垃圾焚烧过程中,苯到氯酚的反应途径亦有可能有相似的反应路径,如图1所示,各基元反应中涉及到的过渡态结构见支持信息图 1.以 Cl2为氯源提供 Cl·取代基取代苯分子上 H,发生两次取代反应依次越过两个较高的势垒形成 1,2-二氯苯(o-DCB),可以发现芳香烃的二氯取代比一氯取代反应势垒高度更大,而放出的反应热降低.o-DCB可能被高温条件下氧化性强的单线态O2直接氧化形成中间体IM1,再经H2还原形成中间体IM2,IM2经分子内H质子的迁移重排、脱水形成产物.同时o-DCB也可能被高能HO·进攻形成IM3或IM4,之后经H⋅、Cl⋅或HO·抽提中间体羟基位H形成2,3-二氯苯酚或 3,4-二氯苯酚,该基元反应为强放热反应.前一种反应路径基元反应普遍势垒高度远高于后一种路径,因此在竞争反应形成最终产物上存在明显劣势.
2.1.2 二氯苯酚形成苯氧自由基 氯代苯酚分子结构相对稳定,之前的研究结果表明 PCDD/Fs可以经过自由基-自由基、自由基-分子、氯酚分子之间反应形成[22].其中苯氧自由基之间的反应是占主导地位,氯代苯氧自由基由于自身不易分解,毒性强,被归类于持久性有机污染物,同时它与其他物质反应活性尤其是氧气反应活性比较低,因此可以进一步作为生成PCDD/Fs的前驱物[23].
生成苯氧自由基(XPRs)是均相反应生成PCDD/Fs的重要步骤之一[22].在高温热解条件下,二氯苯酚最有可能通过热分解反应发生以下反应[24-25]:
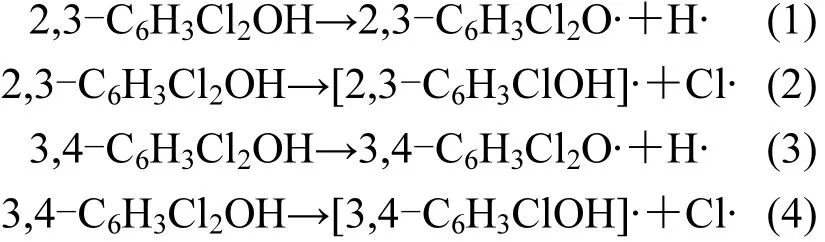
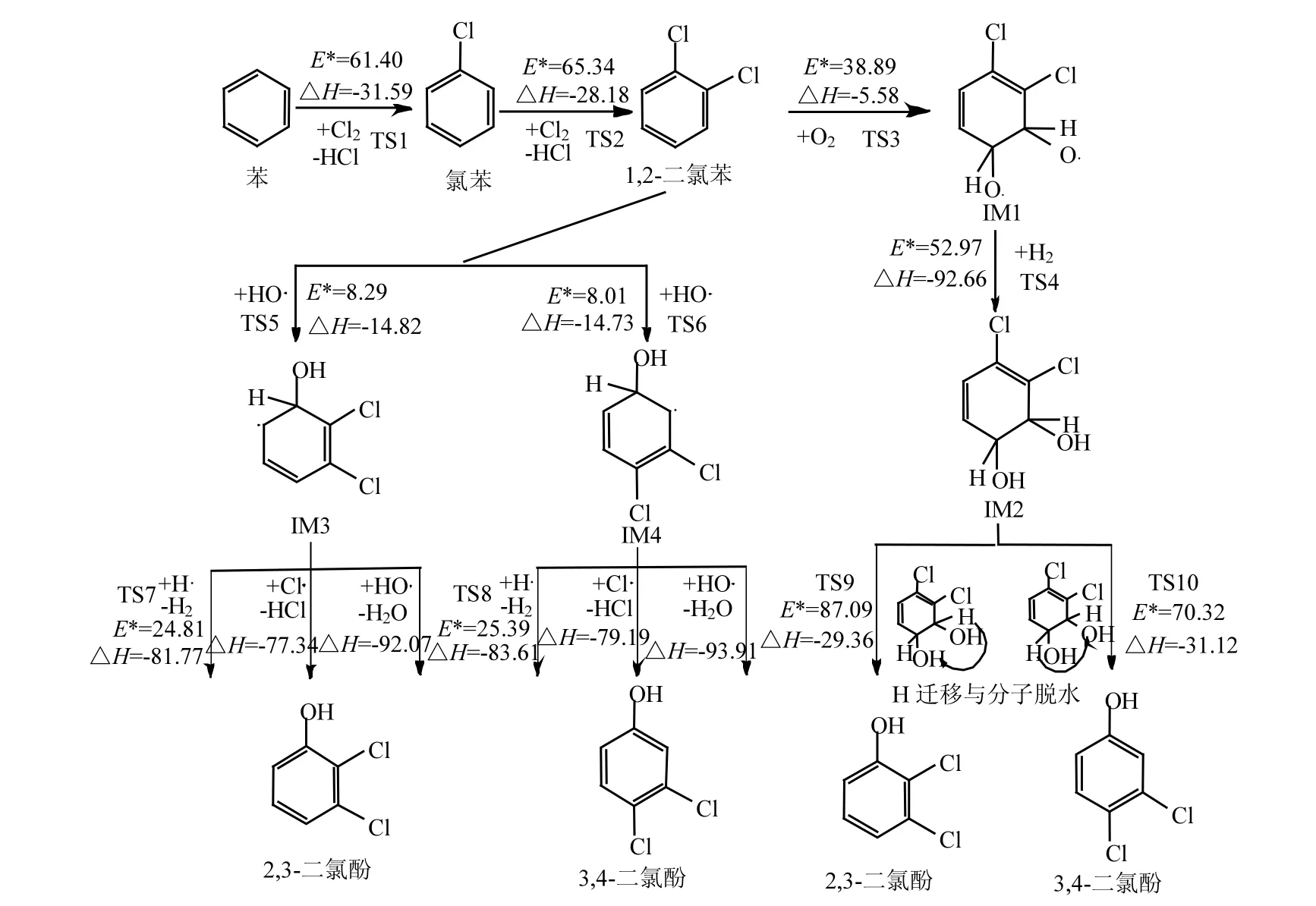
图1 由苯形成2,3-二氯酚及3,4-二氯酚的反应机理Fig.1 Formation of 2,3-dichlorophenol and 3,4-dichlorophenol through the benzene E*:势垒,kcal/mol;△H:反应热,kcal/mol
Xu等[26]在1070~1150K下对于苯酚的动力学模型做了单分子热解模拟,由于苯酚中羟基O-H键作用力弱于芳香环C-H键,因此苯酚分子中H-X最有可能是H-O键分解,化学反应速率常数为 k = 2 .67× 1 016e(-44700K/T)s-1.Ritter(phenol)等[27]在 1070~1028K温度范围下拟合氯苯脱除Cl原子化学反应速率常数为 k(chlorobenzene)=3.0× 1015e(-48100K/T)s-1.Cl脱离芳香环的化学反应速率低于苯酚形成苯氧自由基 H-O键断裂的化学反应速率1个数量级以上,因此氯代苯酚的热分解反应最有可能发生的是苯氧基的 O-H键断裂形成苯氧自由基的过程.在高温以及垃圾焚烧复杂的环境下,苯氧自由基可能经过单分子、双分子或其他形式的基元反应形成,其中单分子反应指酚羟基 O-H键断裂,双分子反应包括复杂环境中高能 H⋅、HO·以及 Cl⋅的进攻等[28].在温度高于900K时,苯氧自由基的形成更倾向于单分子解离;而在温度低于900K时,其他高能原子及自由基团的进攻更容易脱除苯氧基上的H[29].

表1 2,3-DCP及3,4-DCP形成2,3-DCPR及3,4-DCPR的势垒与反应热Table 1 The potential barriers E* and reaction heats △H for the formation of the 2,3-DCPR and 3,4-DCPR from 2,3-DCP and 3,4-DCP through various processess
研究发现C-H键的直接断裂是一个强吸热反应;使用HO·提取二氯酚酚羟基上H的势垒要远小于 H·提取,但放出的反应热较高;尤其说明的是使用Cl·抽提获得的经零点能校正的过渡态总能量,要低于2,3-DCP及3,4-DCP与Cl反应的总能量,因此该反应为无势垒反应.这一计算结果与 Zhang等[10]在 MPWB1K/6-311+G(3df,2p)对Cl提取2,4,6-TCP及2,4-DCP羟基上的H生成2,4,6-TCPR及2,4-DCPR的结论一致.相关反应势垒及反应热见表1.
2.2 由2,3DCPR及3,4DCPR形成PCDD/Fs的过程分析
2.2.1 由2,3-DCPR与3,4-DCPR形成1,2,8,9-TCDD及1,2,7,8-TCDD的机理 基于2,3-DCPR与3,4-DCPR形成1,2,8,9-TCDD及1,2,7,8-TCDD过程主要的基元反应包括:二氯代苯氧自由基之间发生的碳氧耦合二聚化反应、H提取反应、环闭合反应、以及最后分子内H消除反应.其形成机理见图 2,相关过渡态结构见支持信息图3.
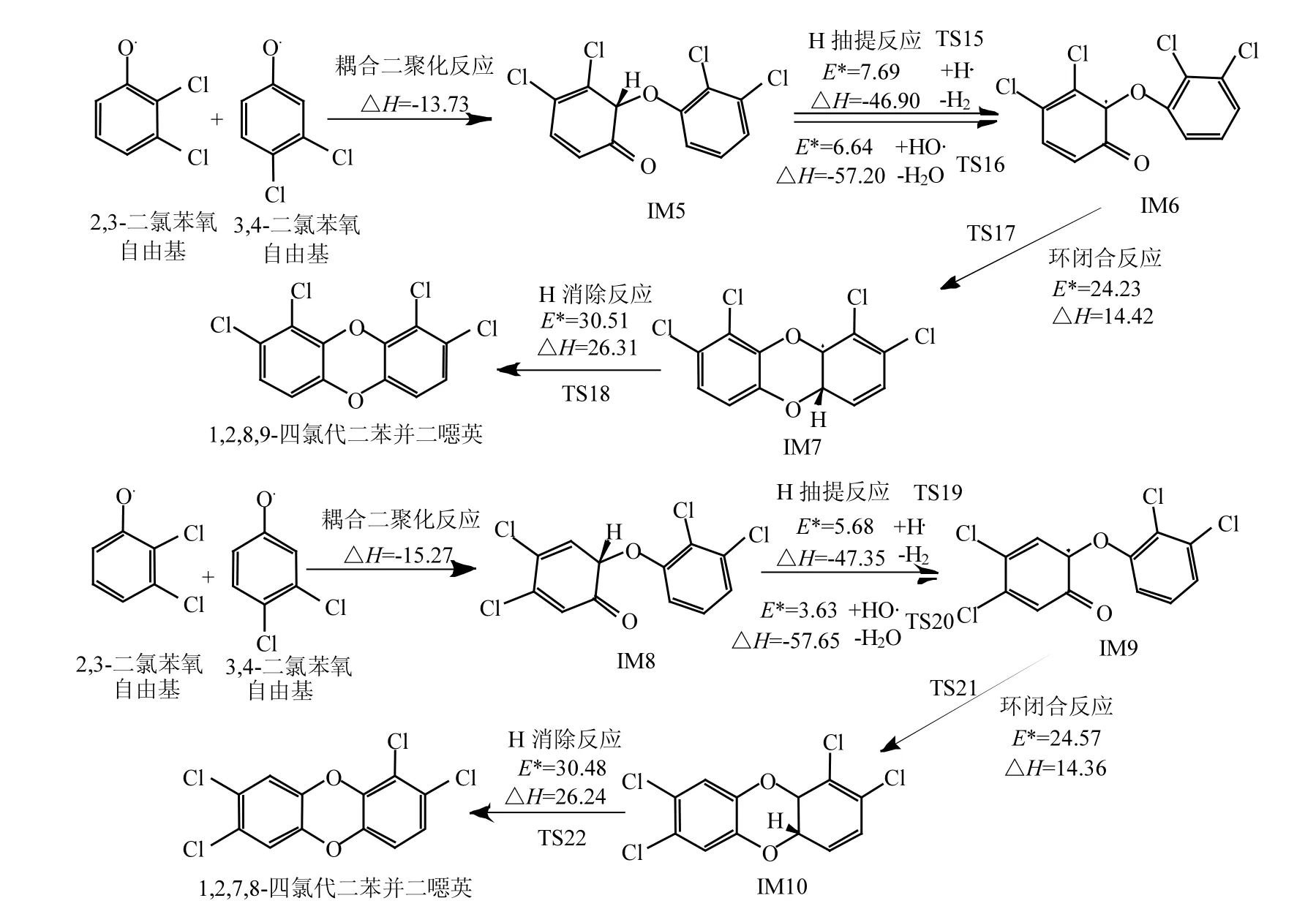
图2 以2,3-DCPR与3,4-DCPR为前驱物形成1,2,8,9-TCDD及1,2,7,8-TCDD的机理Fig.2 1,2,8,9-TCDD and 1,2,7,8-TCDD formation routes from the 2,3-dichlorophenol and 3,4-dichlorophenol precursor E*:势垒,kcal/mol;△H:反应热,kcal/mol
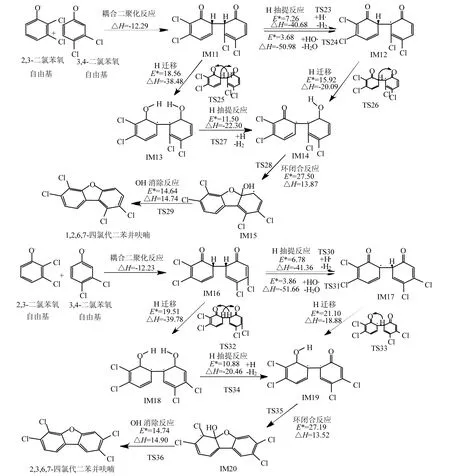
图3 以2,3-DCPR与3,4-DCPR为前驱物形成2,3,6,7-TCDF 及1,2,6,7-TCDF的气相机理Fig.3 2,3,6,7-TCDF and 1,2,6,7-TCDF formation routes from the 2,3-dichlorophenol and 3,4-dichlorophenol precursor E*:势垒,kcal/mol;△H:反应热,kcal/mol
在1,2,8,9-TCDD形成过程的基元反应中,2,3-DCPR与3,4-DCPR的碳氧二聚化耦合反应为无势垒放热反应,反应热为13.73kcal/mol.利用高能H·去提取中间体IM5上H,脱除H2的过程,比HO·自由基去提取H脱除H2O需跨越的势垒高度大,但基元反应放热量少.从中间体IM6经过环闭合反应需要跨越较大的势垒同时吸收一定反应热生成中间体IM7.与上一基元反应相比,从中间体IM7到最终产物1,2,8,9-TCDD需要跨越更高的势垒,达30.51kcal/mol,同时吸收更多的反应热,为本反应形成过程的决速步.1,2,7,8-TCDD与1,2,8,9-TCDD形成过程的基元反应基本相同,只是在最初的碳氧耦合反应时不同碳原子之间发生二聚化反应,形成同分异构中间体IM8,该步基元反应放热量高于1,2,8,9-TCDD基元反应放热.对IM8与IM5的空间结构进行几何结构优化发现,两分子结构芳香环碳氧耦合处 IM8分子C-H键键级为 0.283,IM5分子 C-H键键级为0.209,因此IM8分子C-H键共价键力要强于后者,C-H键更稳定,因此在H提取过程中需要放出更多的热量;同时我们也发现,提取IM8分子中H跨越的势垒高度要高于IM5分子,最可能的原因是邻位卤素原子Cl提高了H提取的活化能,增大了 H·提取的难度.从中间体 IM10至最终产物1,2,7,8-TCDD也是本反应过程的决速步骤,该基元反应需要跨越的势垒以及需要消耗的反应热略小于 1,2,8,9-TCDD 的形成过程,因此在形成1,2,7,8-TCDD时有一定的优势,理论上 1,2,7,8-TCDD的产率要略高于1,2,8,9-TCDD.同时由于两基元反应的势垒与反应热差值较小,因此二者在形成过程中可能存在竞争机制.
2.2.2 由 2,3-DCPR与 3,4-DCPR形成 1,2,6,7-TCDF及2,3,6,7-TCDF的机理 Werber等[30]研究显示,基于氯代苯氧自由基邻位C-C原子的耦合形成中间体二氯代二氧代联苯是多氯联苯并呋喃形成的关键基元反应.由 2,3-DCPR与3,4-DCPR形成1,2,6,7-TCDF及2,3,6,7- TCDF的过程主要包含的基元反应包括:不同苯氧自由基邻位碳原子耦合二聚化反应,H的抽提,单原子或双原子H的迁移重排,环闭合反应,OH消去反应.如图 3所示,1,2,6,7-TCDF的形成过程中,IM11的进一步可能的基元反应包括H的抽提或者双 H 迁移重排,因此有两条反应路径.利用H·以及HO·抽提IM11上的H需要跨越的势垒均远小于双 H 的迁移重排,因此 IM11→IM12比IM11→IM13更加容易发生.从IM13→IM14发生羟基中H的抽提,势垒高度要大于IM11→IM12,但反应热约为后者的一半,说明 IM13中羟基中H提取难度大于IM11中C-H,且H-O键能小于C-H.从 IM14→IM15在反应路径中势垒最高达27.50kcal/mol,同时吸收 13.87kcal/mol反应热,为本过程的控速步骤.IM15→1,2,6,7-TCDF为最终OH消去反应,同样需要吸收反应热.
2,3,6,7-TCDF的形成过程与 1,2,6,7-TCDF相同(图 3,图 4).由 2,3-DCPR 与 3,4-DCPR 经C-C耦合二聚化生成的IM16与IM11互为同分异构体,前者的反应热略高于后者.基元反应IM16→IM17、IM16→IM18、IM18→IM19 跨越的势垒以及反应热与同过程1,2,6,7-TCDF无明显数值差异.IM17→IM19基元反应势垒比IM12→IM14大5.18kcal/mol,可能的原因是IM12苯氧基同侧邻位Cl提高了生成羟基的势垒,但反应热无明显变化.环闭合反应 IM19→IM20也是本反应的决速步骤,势垒高度为27.19kcal/mol,吸收反应热 13.52kcal/mol.最后中间体经过 OH脱除形成2,3,6,7-TCDF.
2.3 速率常数计算
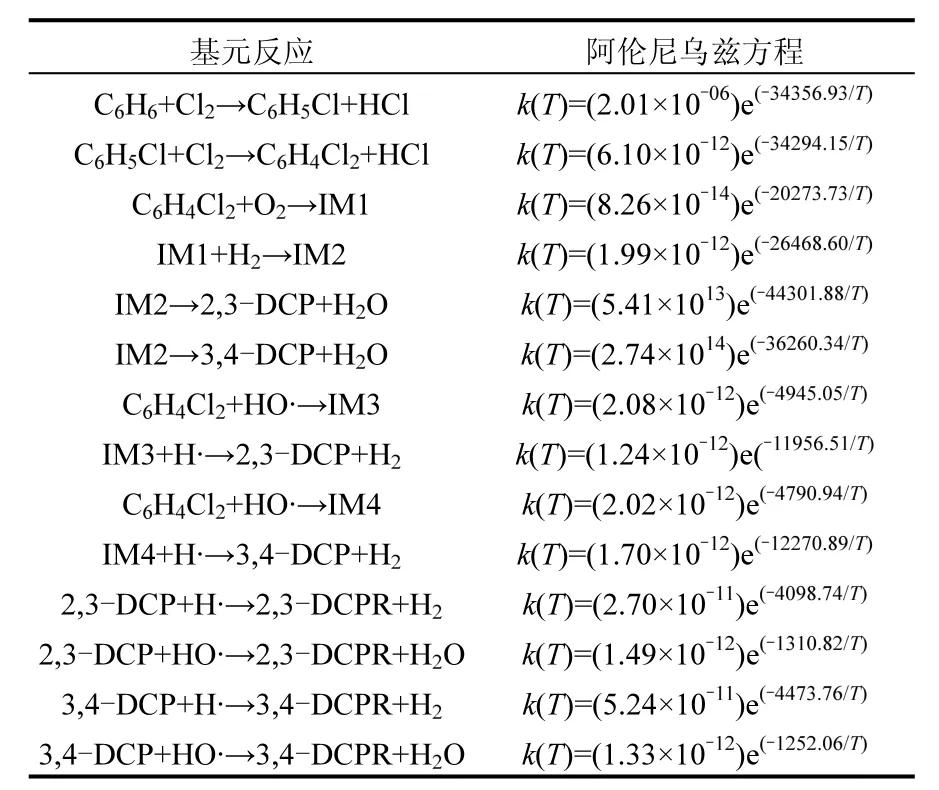
表2 300~1300K温度范围内苯形成苯氧自由基涉及基元反应的Arrhenius方程Table 2 Arrhenius formulas in the formation of phenoxy radical from the benzene over the temperature range of 300~1300K
环境监督与风险决策分析通过建立数学模型研究污染物释放到环境中的潜在结果,PCDD/Fs形成过程中各基元反应阿伦尼乌斯公式中的指前因子、活化能、速率常数是数学模型建立过程中重要的参数[29].为此,基于变分过渡态理论VTST拟合了300~1300K温度范围内的TST速率常数的速率-温度关系式,该温度范围已经涵盖了垃圾焚烧过程中可能涉及到的形成温度.得到各相关过渡态基元反应的阿伦尼乌斯公式,到目前为止,相关文献缺乏直接的相关实验值与理论值.为验证本文拟合公式的准确性,与Gao 等[31]及 Zhang 等[10]在 MPWB1K/ 6-311+G(3df,2p)水平下计算得到的类似基元反应的数据进行对比,并分析在 CVT/SCT拟合的化学反应速率常数,发现相似基元反应的数量级处于同等水平.例如,本文计算得到 2,3-DCP+ H·→2,3-DCPR+H2指前因子为2.70×10-11s-1, Zhang等[10]获得 2,4-DCP+H→2,4-DCPR+H2的指前因子为5.01×10-11s-1.形成 1,2, 8,9-TCDD 环闭合反应IM7→1,2,8,9-TCDD+H·指前因子为 4.05×1013s-1,Zhang等拟合获得2个2,3-DCPR分子形成1,3,6,8-TCDD、1,3,7,9-TCDD各基元反应,相同闭环反应的指前因子分别为3.17×1013s, 2.96×1013s-1,因此本文计算数据可靠.由苯两阶段生成PCDD/Fs涉及的基元反应并拟合获得的阿伦尼乌斯公式如表2,表3所示.

表3 300~1300K温度范围内2,3-DCPR和3,4-DCPR为前驱物形成PCDD/Fs涉及基元反应的Arrhenius方程Table 3 Arrhenius formulas in the formation of PCDD/Fs from the 2,3-DCPR and 3,4-DCPR precursor over the temperature range of 300~1300K
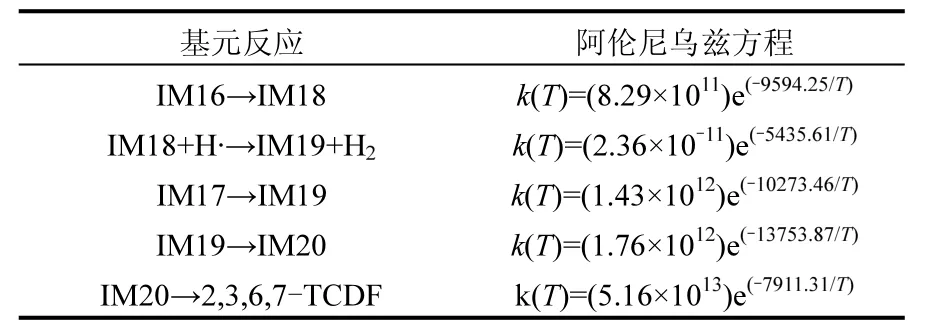
续表3
3 结论
3.1 基于密度泛函理论研究了以苯为前驱物分两阶段形成PCDD/Fs的均相反应机理.在第一阶段苯的氯化、氧化各基元反应中,氯化取代反应势垒较高,羟基自由基进攻二氯苯进而形成二氯酚过程势垒明显低于氧气直接氧化氯苯经加氢分子内脱水反应.
3.2 苯氧自由基的之间的碳氧耦合二聚化反应比碳碳耦合二聚化释放的反应热多.邻位Cl能够提高芳香烃环H的抽取势垒,也会增加邻位羟基反应势垒,降低了分子反应活性.
3.3 环闭合基元反应在第二阶段的反应中需要越过的势垒最大,是形成 1,2,7,8-TCDD、1,2,8,9-TCDD、1,2,6,7-TCDF、2,3,6,7-TCDF的决速步.
3.4 苯为反应活性较低的二噁英前驱物,以苯为前驱物生成PCDD/Fs化学反应速率的快慢主要取决于苯的氯化基元反应.
[1]Schoonenboom M H, Tromp P C, Olie K. The formation of coplanar PCBs, PCDDs and PCDFs in a fly ash model system [J].Chemosphere, 1995,30(7):1341-1349.
[2]Huang H, Buekens A. On the mechanisms of dioxin formation in combustion processes [J]. Chemosphere, 1995,31(9):4099-4117.
[3]Addink R, Olie K. Mechanisms of Formation and Destruction of Polychlorinated Dibenzo-p-dioxins and Dibenzofurans in Heterogeneous Systems [J]. Environmental Science &Technology, 1995,29(6):1425-1435.
[4]Shi X, Zhang R, Zhang H, et al. Influence of water on the homogeneous gas-phase formation mechanism of polyhalogenated dioxins/furans from chlorinated/brominated phenols as precursors [J]. Chemosphere, 2015,137:142-148.
[5]陈 彤.城市生活垃圾焚烧过程中二噁英的形成机理及控制技术研究 [D]. 杭州:浙江大学, 2006.
[6]Ghorishi S B, Altwicker E R. Rapid formation of polychlorinated dioxins/furans during the heterogeneous combustion of 1,2-dichlorobenzene and 2, 4-dichlorophenol [J]. Chemosphere,1996,32(1):133-144.
[7]Altwicker E R. Formation of PCDDF in municipal solid waste incinerators: laboratory and modeling studies [J]. Journal of Hazardous Materials, 1996,47(1-3):137-161.
[8]Ryan S P, Altwicker E R. The formation of polychlorinated dibenzo-p-dioxins/dibenzofurans from carbon model mixtures containing ferrous chloride. [J]. Chemosphere, 2000,40(9-11):1009-1014.
[9]Schoonenboom M H, Baints E, Olie K, et al. The formation of chlorinated compounds from benzene in a fly ash model system [J].Toxicological & Environmental Chemistry, 1995,52(52):1-11.
[10]Zhang Q, Yu W, Zhang R, et al. Quantum chemical and kinetic study on dioxin formation from the 2,4,6-TCP and 2,4-DCP precursors [J]. Environmental Science & Technology, 2010,44(9):3395-3403.
[11]Dharmarathne N K, Mackie J C, Kennedy E M, et al. Gas phase pyrolysis of endosulfan and formation of dioxin precursors of polychlorinated dibenzo-p-dioxins and dibenzofurans (PCDD/F)[J]. Proceedings of the Combustion Institute, 2016,36(1):1119-1127.
[12]Saeed A, Altarawneh M, Dlugogorski B Z. Formation of mixed halogenated dibenzo-p-dioxins and dibenzofurans (PXDD/Fs) [J].Chemosphere, 2015,137:149-156.
[13]Yang X, Liu H, Hou H, et al. Studies of thermodynamic properties and relative stability of a series of polyfluorinated dibenzo-p-dioxins by density functional theory. [J]. Journal of Hazardous Materials, 2010,181(1-3):969.
[14]Qu R, Liu H, Zhang Q, et al. The effect of hydroxyl groups on the stability and thermodynamic properties of polyhydroxylated xanthones as calculated by density functional theory [J].Thermochimica Acta, 2012,527(2):99-111.
[15]Lee C, Yang W, Parr R G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density [J]. Phys. Rev. B Condens. Matter, 1988,37(2):785-789.
[16]Truhlar D G, Garrett B C, Hipes P G, et al. Test of variational transition state theory against accurate quantal results for a reaction with very large reaction-path curvature and a low barrier[J]. Journal of Chemical Physics, 1984,81(8):3542-3545.
[17]Dar T, Shah K, Moghtaderi B, et al. Formation of persistent organic pollutants from 2,4,5-trichlorothiophenol combustion: a density functional theory investigation [J]. Journal of Molecular Modeling, 2016,22(6):1471-1475.
[18]Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09,Revision A. 1. Wallingford, CT: Gaussian [Z]. Inc, 2009.
[19]Yamanaka H, Hamada R, Nibuta H, et al. Gas-phase catalytic oxidation of benzene over Cu-supported ZSM-5catalysts: an attempt of one-step production of phenol [J]. Journal of Molecular Catalysis A: Chemical, 2002,178(1):89-95.
[20]Kato C N, Hasegawa M, Sato T, et al. Microporous dinuclear copper (II) trans-1, 4-cyclohexanedicarboxylate: heterogeneous oxidation catalysis with hydrogen peroxide and X-ray powder structure of peroxo copper (II) intermediate [J]. Journal of Catalysis, 2005,230(1):226-236.
[21]王伟涛,姚 敏,马养民,等.氧气直接氧化苯制备苯酚 [J]. 化学进展, 2014,26(10):1665-1672.
[22]Dar T, Altarawneh M, Dlugogorski B Z. Theoretical study in the dimerisation of 2-chlrothiophenol/2-chlorothiophenoxy:Precursors to PCDT/TA [J]. Organohalogen Compounds, 2012,74:657-660.
[23]Ritter E R, Bozzelli J W, Dean A M. Kinetic study on thermal decomposition of chlorobenzene diluted in hydrogen [J].Cheminform, 1990,21(27):2493-2504.
[24]Evans C S, Dellinger B. Mechanisms of dioxin formation from the high-temperature pyrolysis of 2-chlorophenol [J].Environmental Science & Technology, 2003,37(7):1325-1330.
[25]Altarawneh M, Dlugogorski B Z, Kennedy E M, et al.Mechanisms for formation, chlorination, dechlorination and destruction of polychlorinated dibenzo-p-dioxins and dibenzofurans (PCDD/Fs) [J]. Progress in Energy and Combustion Science, 2009,35(3):245-274.
[26]Xu Z F, Lin M C. Ab initio kinetics for the unimolecular reaction C6H5OH→CO+C5H6[J]. The Journal of Physical Chemistry A,2006,110(4):1672-1677.
[27]Ritter E R, Bozzelli J W, Dean A M. Kinetic study on thermal decomposition of chlorobenzene diluted in hydrogen [J].Cheminform, 1990,21(27):2493-2504.
[28]Evans C S, Dellinger B. Mechanisms of dioxin formation from the high-temperature oxidation of 2-chlorophenol. [J].Environmental Science & Technology, 2005,39(1):122-127.
[29]郁万妮.以卤代苯酚为前体物的二噁英气相形成机理研究 [D].济南:山东大学, 2013.
[30]Weber R, Hagenmaier H. Mechanism of the formation of polychlorinated dibenzo-p-dioxins and dibenzofurans from chlorophenols in gas phase reactions [J]. Chemosphere, 1999,38(3):529-549.
[31]Gao R, Xu F, Li S, et al. Formation of bromophenoxy radicals from complete series reactions of bromophenols with H and OH radicals. [J]. Chemosphere, 2013,92(4):382-390.
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
大学物理(2022年1期)2022-01-13
灾害医学与救援(电子版)(2018年1期)2018-06-05
电脑知识与技术(2018年3期)2018-03-21
中国资源综合利用(2017年4期)2018-01-22
哈尔滨理工大学学报(2017年1期)2017-04-08
电子制作(2017年19期)2017-02-02
科技视界(2016年24期)2016-10-11
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
郑州大学学报(理学版)(2014年4期)2014-03-01
