
过渡金属-膦配体在催化反应中的应用
2017-12-25 03:28厉嘉云彭家建
杭州师范大学学报(自然科学版) 2017年6期
杨 闯,厉嘉云,彭家建,白 赢
(杭州师范大学有机硅化学及材料技术教育部重点实验室,浙江 杭州 311121)
过渡金属-膦配体在催化反应中的应用
杨 闯,厉嘉云,彭家建,白 赢
(杭州师范大学有机硅化学及材料技术教育部重点实验室,浙江 杭州 311121)
膦配体是均相过渡金属催化反应中使用最广的一类配体,其立体效应和电子效应对过渡金属催化反应的影响很大.为提高反应的转化率和产物的选择性,设计和修饰膦配体成为过渡金属催化反应研究的重点.文章综述了过渡金属非手性膦配合物在催化反应中的研究进展.
膦配体;过渡金属配合物;催化剂
为提高均相过渡金属催化反应的转化率和目标产物选择性,如何设计和修饰膦配体得到了广泛研究.过渡金属膦配合物在催化反应中的应用主要包括加氢反应、N-甲基化反应、甲酰化反应、偶联反应、烯丙基化反应、乙烯齐聚反应[1-2]、Michael加成[3-4]、硅氢加成反应等.

1 膦配体在过渡金属配位催化反应中的应用
1.1 过渡金属膦配合物在催化加氢反应中的应用



图1 cis-[RuCl2(dcype)(1a)]催化加氢反应Fig. 1 Hydrogenation catalyzed with cis-[RuCl2(dcype)(1a)]
至少含一个甲硅烷基的配体与过渡金属形成的配合物得到研究者们的广泛关注,主要因为该类膦配体其σ给电子能力较强.2015年,Komuro等[6]报道含有二甲基硅基的膦配体2与Ru(H)Cl(PPh3)3反应得到配合物3(图2),并证实配合物3和配合物4的结构在一定条件下是互变的.将配合物3在60 ℃且C6D6作为反应溶剂的条件下催化苯乙烯的加氢反应,转化率接近100%.其催化4-甲氧基苯乙烯加氢反应转化率为91%,4-乙基苯甲醚的选择性为95%;催化3,3-二甲基-1-丁烯加氢反应转化率为88%,2,2-二甲基丁烷的选择性接近100%.

图2 Ru(H)Cl(PPh3)3/2催化加氢反应Fig. 2 Hydrogenation catalyzed with Ru(H)Cl(PPh3)3/2
2016年,Rodrigues等[7]合成5种含P和吡啶基的钌配合物5a-5e,统称为mer-[RuCl3(dppb)(N)](N为吡啶(5a),对甲基吡啶(5b),对乙烯基吡啶(5c),对叔丁基吡啶(5d),对苯基吡啶(5e)).当mer-[RuCl3(dppb)(5c)]作为反应催化剂(图3),底物分别为环己烯、十一醛、环己基甲醛时,反应转化率分别达96%,93%,73%.
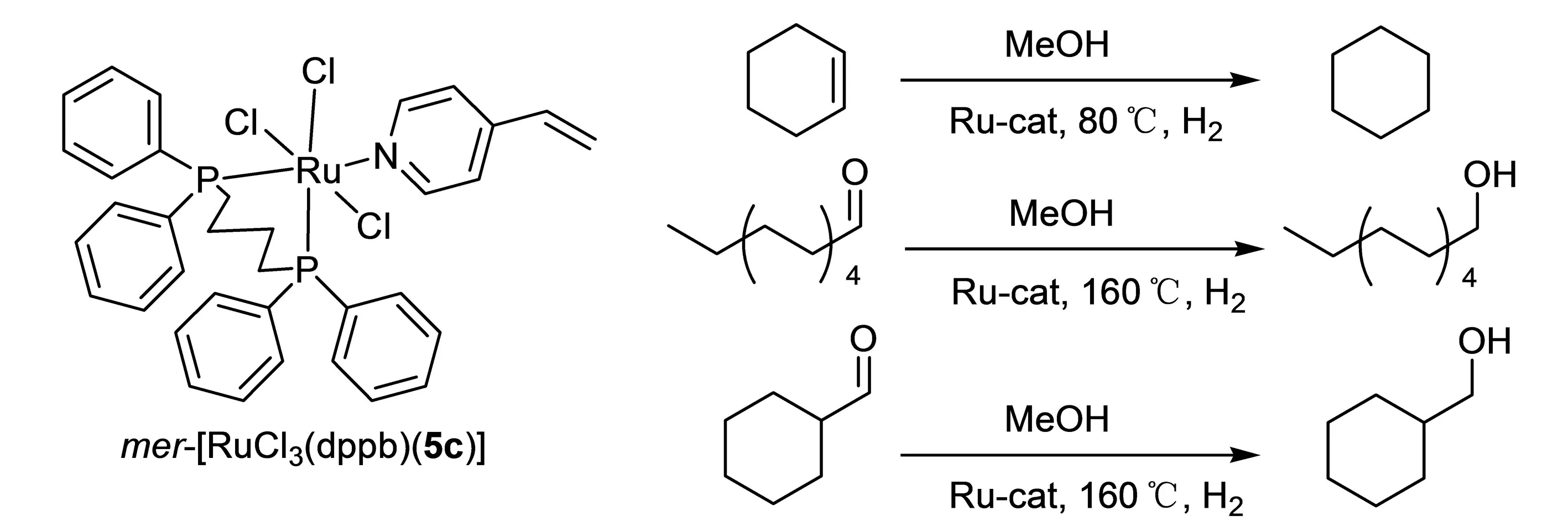
图3 mer-[RuCl3(dppb)(5c)]催化加氢反应Fig. 3 Hydrogenation catalyzed with mer-[RuCl3(dppb)(5c)]
1.2 过渡金属膦配合物在催化构建N—C键反应中的应用
使用廉价且无毒的CO2作为C1源制备附加值大的化学物质具有重要意义.CO2转化为甲酸、甲醇(甲氧基)、甲烷等已得到了广泛的研究.但应用CO2作为胺的甲基化试剂还鲜有报道.
Jessop等[8]报道了仲胺在超临界CO2中的反应(图4),以钌的络合物RuH2[P(CH3)3]4和RuCl2[P(CH3)3]4催化二甲胺得到N,N-二甲基甲酰胺,反应TOF可达8 000 h-1.由于超临界二氧化碳具有弱的溶剂化作用,对氢气具有很好的混合性,同时对过渡金属配合物有很好的溶解度,使体系为高分散的均相体系,有利于反应的进行.

图4 RuCl2[P(CH3)3]4催化二甲胺的甲基化反应Fig. 4 Methylation of dimethylamine with RuCl2[P(CH3)3]4
2013年,Li等[9]利用多种膦配体(图5,6a-6f)与钌化合物[RuCl2(DMSO)4]形成配合物,催化CO2与不同类型胺的反应.以[RuCl2(DMSO)4]/nBuPAd2(Ad=金刚烷基)作催化剂、PhSiH3为硅烷、110 ℃条件下反应16 h,催化N-甲基苯胺与CO2的甲基化反应所得N,N-二甲基苯胺的产率为92%,而不加配体时产率仅为70%.其在最佳条件下用来催化取代苯胺类的N-甲基化反应,均取得了较好的催化效果.

图5 N-甲基苯胺的甲基化反应Fig. 5 Methylation of N-methylaniline
2015年,Dabbawala等[10]合成了7-15膦配体(图6),将其与RuCl3·3H2O和RuCl2(PPh3)3得到的配合物用以催化二乙胺的甲酰化反应.未加入膦配体时二乙胺转化率只有1%,但加入适当膦配体后即使CO2和H2在较低的压力下也可提高二乙胺的转化率.采用三齿膦配体15时的反应TOF高于采用位阻较大的单齿膦配体的TOF,但低于PPh3和双齿膦配体的.而在采用1,2-双(二苯基膦基)苯配体14时,可以得到最好催化效果:反应TON最高可达2 475,反应转化率达99%,N,N-二乙基甲酰胺反应选择性达90%以上.

图6 二乙胺的甲酰化反应Fig. 6 Formylation of Diethylamine
1.3 过渡金属膦配合物在催化氢甲酰化反应中的应用
烯烃氢甲酰化反应是制备醛的一种重要方法.Brown等[11]发现铑催化剂中加入膦配体可以催化氢甲酰化反应,且反应能在较低温度和压力下完成.其中,双膦配体具有特殊的立体效应和电子效应,其与铑形成的配合物对提高烯烃氢甲酰化反应的选择性和反应活性有极大的作用.
2002年,Slot等[12]合成一种新颖的含有N-吡咯基的双齿膦配体16,18(图7).将配体16,18及含苯氧基膦配体17,19与Rh(acac)(CO)2形成的配合物催化1-辛烯的氢甲酰化,发现16,18的铑配合物得到的反应产率较高,直链醛的选择性也较高,其中18的铑配合物得到直链醛的选择性可达92%.通过比较18,19的铑配合物催化1-辛烯的氢甲酰化反应,可发现增加配体的吸电子基团和改变配体的位阻,能极大提高反应转化率和目标产物选择性.
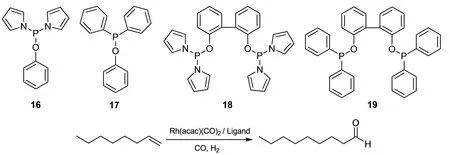
图7 1-辛烯的氢甲酰化反应Fig. 7 Hydroformylation of 1-octene

图8 内烯烃的氢甲酰化反应Fig. 8 Hydroformylation of Internal olefin
关于末端烯烃的氢甲酰化反应已得到较广泛研究,但内烯烃则较少文献报道.Yu 等[13]设计合成了一种较新颖且含有4个P的配体20(图8),其吡咯基团吸电子效应和P本身具有的强螯合能力相结合,(R=H)与过渡金属形成的配合物可显著提高内烯烃氢甲酰化反应生成直链醛的速率.催化2-辛烯和2-己烯的氢甲酰化反应得到直链醛的选择性为98.1%和99.2%.
Tijani 等[14]研究了以Rh(acac)(CO)2和含亚磷酸结构的配体(如21,图9)形成的配合物催化苯乙烯氢甲酰化的反应,转化率可达97%,直链醛和支链醛选择性为44%,56%.催化2-戊烯和2-苯丙烯的转化率则较低,而催化亚甲基环戊烷、3,3-二甲基-1-丁烯的氢甲酰化反应的转化率分别达80%,83%,相应的直链醛选择性均高达98%.
1.4 过渡金属膦配合物在催化偶联反应中的应用
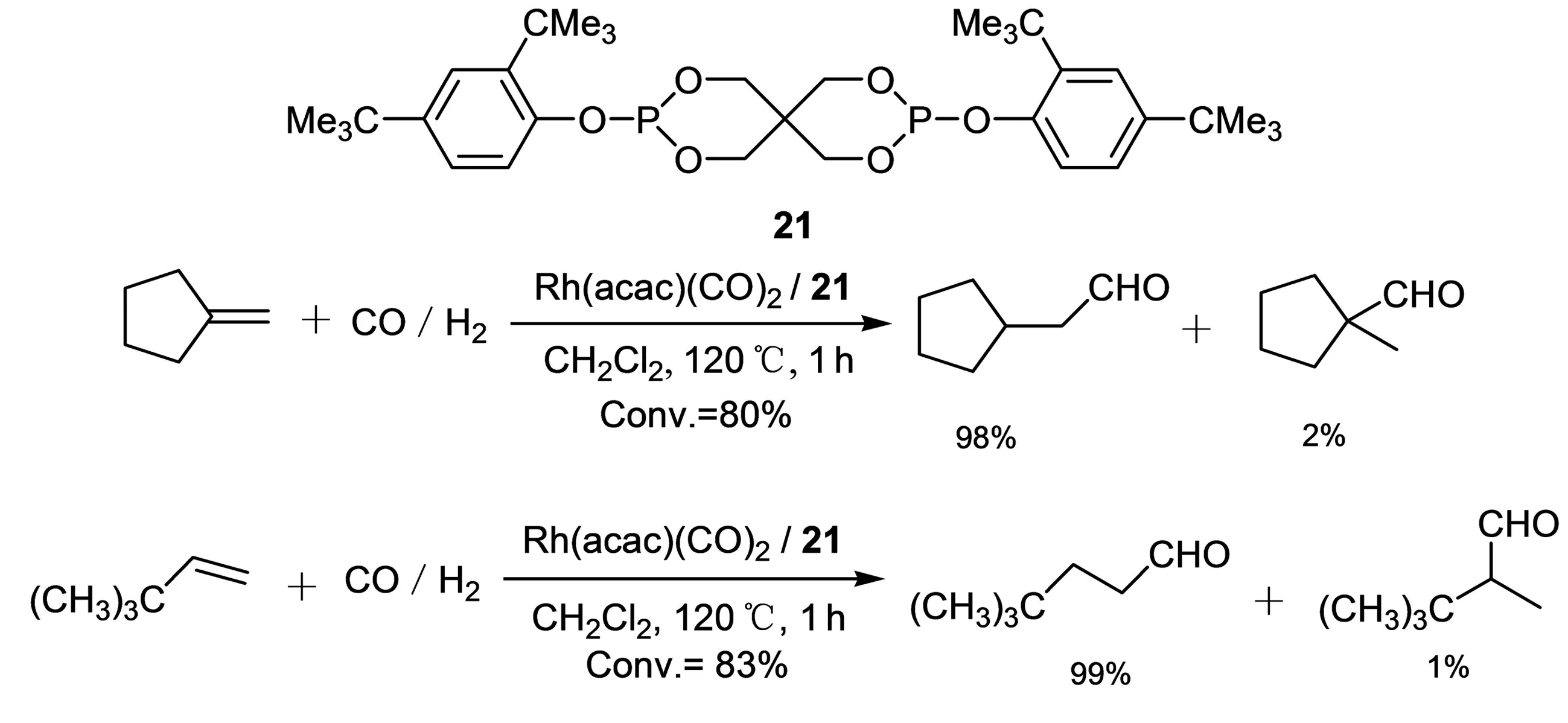
图9 烯烃的氢甲酰化反应Fig. 9 Hydroformylation of alkene
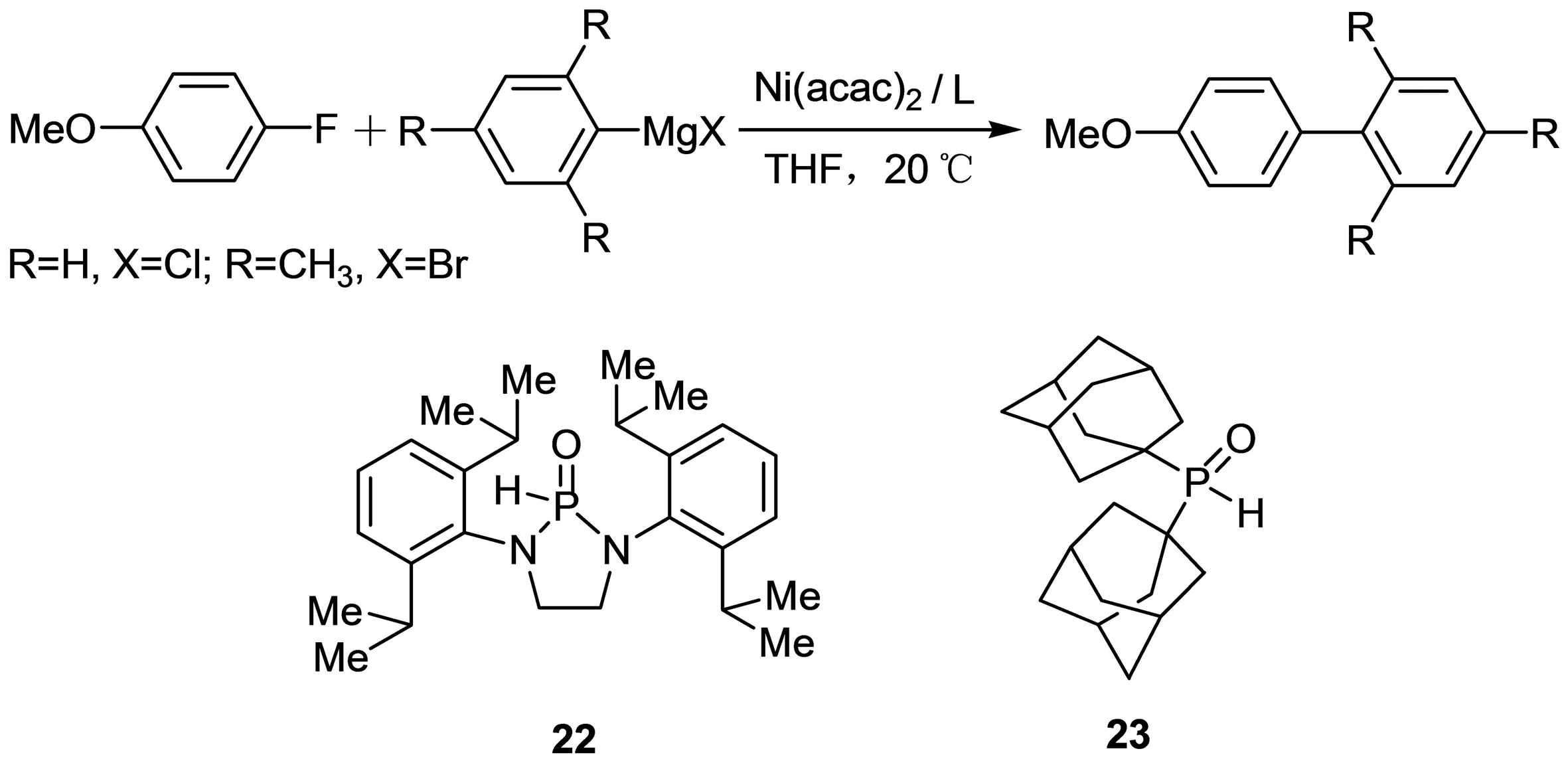
图10 Ni(acac)2/L催化C—C偶联加氢反应Fig. 10 C—C coupling catalyzed with Ni(acac)2/L
过渡金属催化的偶联反应是制备化合物的一种重要方法.而含膦配体过渡金属配合物应用于大多数偶联反应,如Kumada-Corriu反应[15-16]、Stille反应[17]、Suzuki反应[18]、Hiyama反应[19-20]、Negishi反应[21-22]以及端炔作为亲核试剂的Sonogashira反应[23-24]和端烯作为亲核试剂的Heck反应[25-26].
2005年,Ackermann等[27]使用Ni(acac)2与一系列含膦配体等摩尔量混合后催化一系列取代氟苯、取代氯苯与芳基格氏试剂的C—C偶联反应(图10),发现使用22,23配体的催化体系的活性较好,同时,用对甲氧基氟苯作为反应底物,偶联产物产率可以达到80%以上.
Martin等[28]合成一系列具有联苯结构的配体24-26(图11),与Pd(dba)2形成配合物催化4-甲基苯基溴化镁与碘苯的Kumada-Corriu交叉偶联反应,与常规的PCy3,PtBu3,PPh3等膦配体相比,采用联苯式配体得到目标产物的产率普遍较高,其中采用24,25,26时的产率分别为82%,93%,98%.
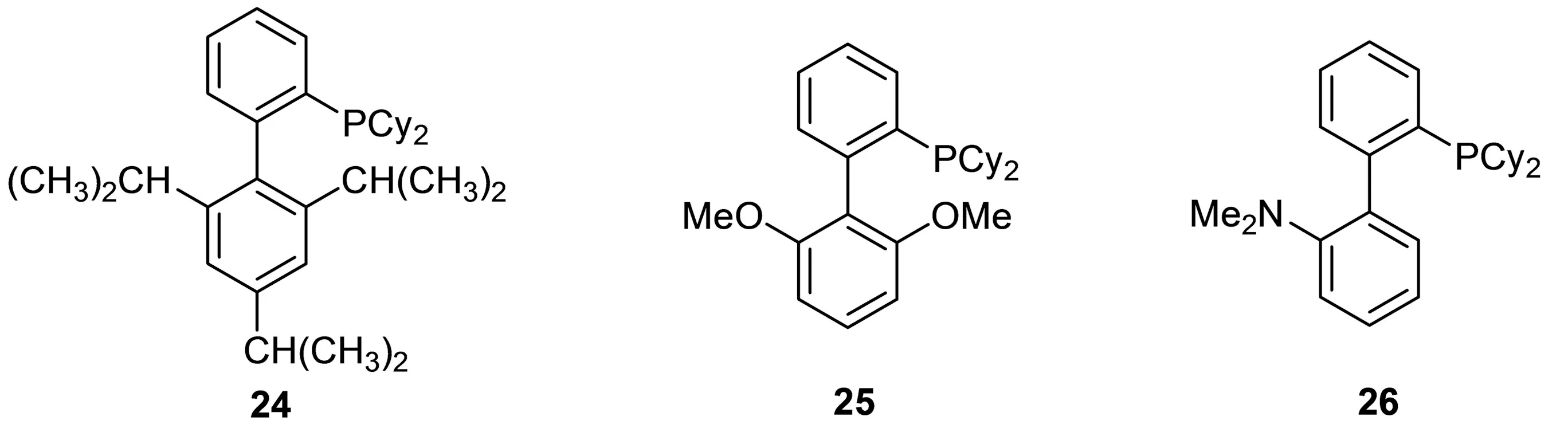
图11 配体24,25,26的结构式Fig. 11 Structure of ligands 24, 25, 26
Biscoe等[29]用25配体与Pd试剂形成配合物在室温下催化PhCl和胺类的C—N偶联反应(图12),得到较高的产率,其中27和28是催化反应过程中关键的中间体.他们通过P谱以及DFT理论计算提出了该催化循环机理:首先钯-膦配合物与底物氯化苯作用,再与底物胺作用生成中间体28,在碱的作用下脱去一分子HX,最后得到产物和原始钯-膦催化剂.当催化氯苯与苯胺的偶联反应时,目标产物二苯胺的选择性达到99%以上.

图12 [Pd]/25催化C—N偶联反应Fig. 12 C—N coupling catalyzed with [Pd]/25

图13 Pd2(dba)3/29催化的C—O偶联反应Fig. 13 C—O coupling catalyzed with Pd2(dba)3/29
Anderson等[30]将具有联苯结构的膦配体29与Pd2(dba)3得到的催化剂采用一锅法来催化芳卤化合物以及卤代物的C—O偶联反应(图13),产物醚收率达到80%以上.该催化反应是通过卤代芳烃、酚类化合物或脂肪族醇合成二芳基醚和烷基芳基醚等产物的一种有效方法.
2008年,Zhang等[31]将30与[PdCl2(PhCN)2]在摩尔比为1∶1的条件下制备了[Pd]/30催化剂(图14).在25 ℃下,催化邻碘苯甲酸乙酯与环己基氯化锌的Negishi偶联反应,11 h后反应完全,产物的收率为99%,反应的TOF达1 000 s-1.同时,用[Pd]/30催化正十二烷基氯化锌和邻碘苯甲酸乙酯的偶联反应也得到了较好的催化效果.

图14 [Pd]/30催化Negishi偶联反应Fig. 14 Negishi coupling catalyzed with [Pd]/30
以Pd(0)作催化剂,芳基卤化物、苯甲酰氯或烯基卤化物和末端烯烃发生C—C偶联的反应称Heck反应,该反应官能团适应性和收率相对较好.Motswaninyana等[32]合成了一种亚胺膦配体31(图15),在CH3CN作反应溶剂、Et3N作碱且回流条件下,用[Pd]/31催化碘代苯和丙烯酸甲酯的C—C偶联反应,反应24 h的转化率为90%,选择性为85%.而在室温下反应8 h后,产率为68%;反应24 h后产率达到75%,反式产物的选择性达到85%.室温下反应的转化率和反应选择性高的原因主要在于亚胺膦配体的半配位效应.
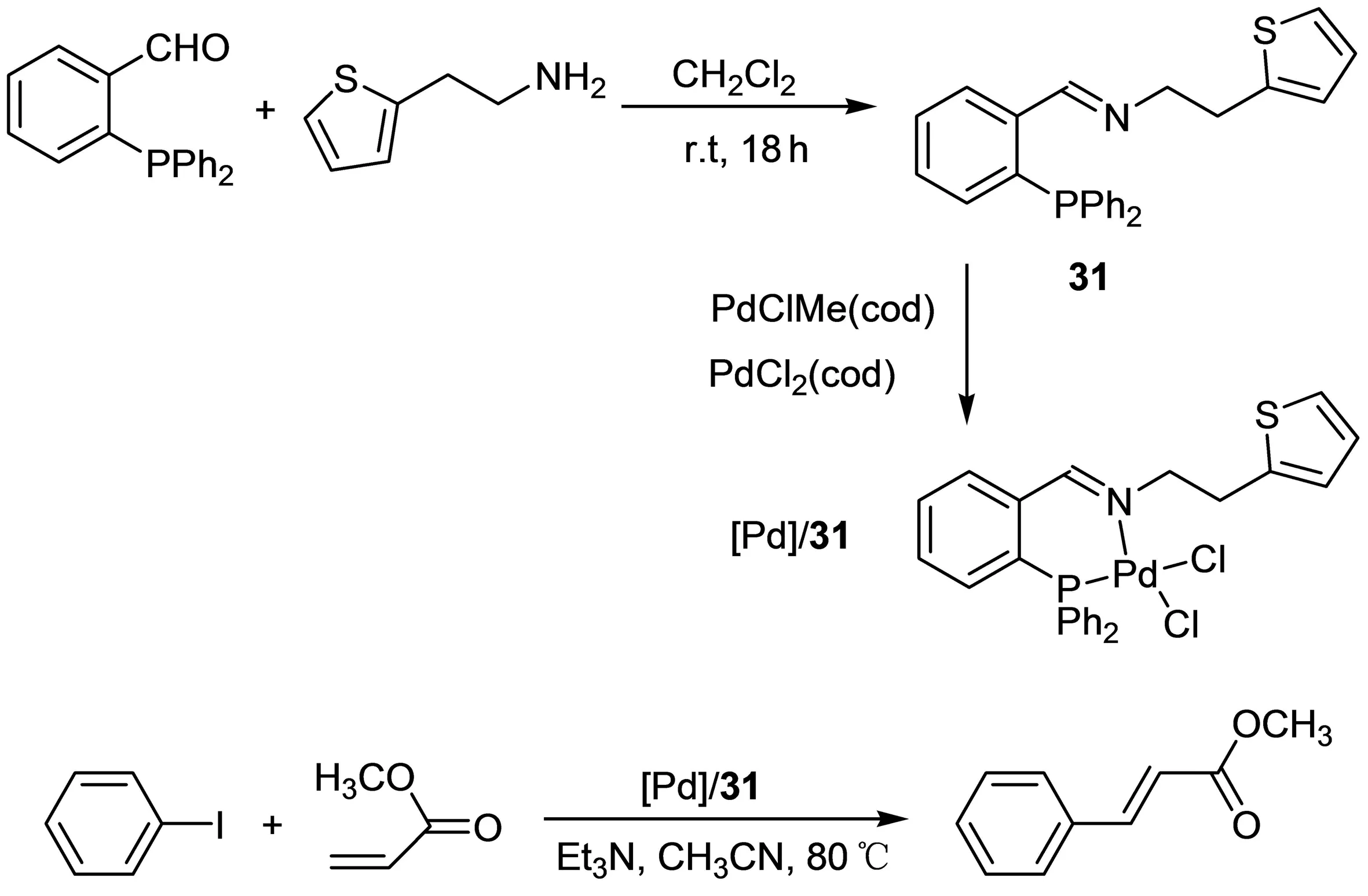
图15 [Pd]/31催化C—C偶联反应Fig. 15 C—C coupling catalyzed with [Pd]/31
与其他偶联反应采用有机金属试剂不同,Suzuki偶联反应使用有机硼试剂,反应后产物较易纯化,且其对水不敏感,同时适用于多种官能团(包括羧基、醛基、氰基等).2000年,Bedford等[33]合成了钯配合物32,33,在K2CO3为碱且THF为溶剂的情况下,分别用来催化4-溴苯乙酮、4-溴苯甲醚和PhB(OH)2的偶联反应(图16).当32,33用量为反应底物的0.001%时,33的催化效果(转化率为92%)明显优于32的催化效果(转化率为59%).
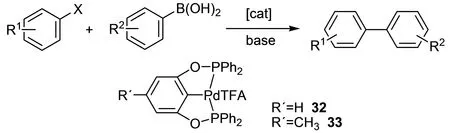
图16 32,33催化Suzuki偶联反应Fig. 16 Suzuki coupling catalyzed with 32,33
1.5 过渡金属膦配合物在催化烯丙基化反应中的应用
2016年,Koschker等[34]报道了铑化合物催化端炔的烯丙基化反应(图17).当[Rh(COD)Cl]2/34配合物(其中COD为1,5-环辛二烯)在1,2-二氯乙烷作反应溶剂,70 ℃下催化1-辛炔与苯甲酸的烯丙基化反应18 h时,反应转化率为80%,支链产物占比为94%.同时,发现[Rh(COD)Cl]2与其他膦配体形成的配合物在催化炔基酸的内酯化以及端炔与硫醇、芳基肼、咪唑等的烯丙基化反应时,均取得了较好的催化效果.

图17 [Rh(COD)Cl]2/34催化烯丙基化反应Fig. 17 Allylation catalyzed with [Rh(COD)Cl]2/34
Lee等[35]研究了氟苯与含有苯丙烯盐基的物质的烯丙基化反应(图18),在Pd(OAc)2作催化剂,P(t-Bu)2(2-OMeC6H4)作为膦配体,Cs2CO3和AgOPiv的作用下,产物为单一的线性产物(E)-烯丙基化氟苯,产率达82%.当无该膦配体时,产率低于5%.在该反应条件下,考察了其他含有烯丙基物质作为底物的催化反应,催化效果均较理想.而当PPh3,PCy3,P(t-Bu)3,PAd2Bu,PCy2Ph等常规膦配体作为催化反应配体时,相应的反应产率仅为5%,16%,14%,17%,22%.

图18 [Pd]/P(t-Bu)2(2-OMeC6H4)催化烯丙基化反应Fig. 18 Allylation catalyzed with [Pd]/P(t-Bu)2(2-OMeC6H4)
1.6 过渡金属膦配合物在催化硅氢加成反应中的应用
Niyomura等[36]将类似碗状的膦配体35与铑化合物形成的配合物用来催化环己酮和二甲基苯基硅烷的硅氢加成反应(图19).当苯为溶剂且n(35)∶n(Rh)=2∶1时,催化效果最好,底物环己酮转化率为97%.与PPh3,PEt3,P(t-Bu)3,PCy3等常规膦配体相比,35膦配体参与的催化硅氢加成反应的效果较好,可能因为该配体空间结构类似碗状,具有一定空间位阻却又不至于位阻过大,不像P(t-Bu)3那样P与相邻原子太过于接近.同时,该膦配体参与催化邻甲基苯甲醛、N-苄烯苯胺、1-己烯与二甲基苯基硅烷的硅氢加成反应的反应产率分别为90%,88%,93%.

图19 [Rh(C2H4)2Cl]2/35催化硅氢加成反应Fig. 19 Hydrosilylation catalyzed with [Rh(C2H4)2Cl]2/35
2008年,Ochida课题组[37]合成了膦配体36(图20).当36和[RhCl(C2H4)2]2摩尔比为1∶1、苯作溶剂且常温下催化邻甲基环己酮、苯乙酮、3-戊酮、4-庚酮、二异丙基酮与二甲基苯基硅烷的硅氢加成反应,转化率分别为100%,93%,100%,83%,72%,显示了该配体与[RhCl(C2H4)2]2形成的配合物非常适合于催化酮类与二甲基苯基硅烷的硅氢加成反应.但在硅烷为Et3SiH时,环己酮和二异丙基酮的转化率极低,甚至环己酮与(t-Bu)Me2SiH几乎不反应.
Saito等[38]合成了一种新型的含卟啉锌的膦配体37(图21),将其与[RhCl(COD)]2化合物配位并应用于催化酮与二苯基氢硅烷的硅氢加成反应.在THF作反应溶剂,n(37)∶n(Rh)=2∶1且室温下反应24 h时,得到苯乙醇(硅氢加成反应完再酸解)转化率为53%.当用对氯苯乙酮、对甲氧基苯乙酮作为底物时,相应产物的产率分别为82%,18%.与不加配体的反应进行比较,该配体可大大提高苯乙酮的转化率,尤其是对氯苯乙酮作为反应底物时,产率最高.
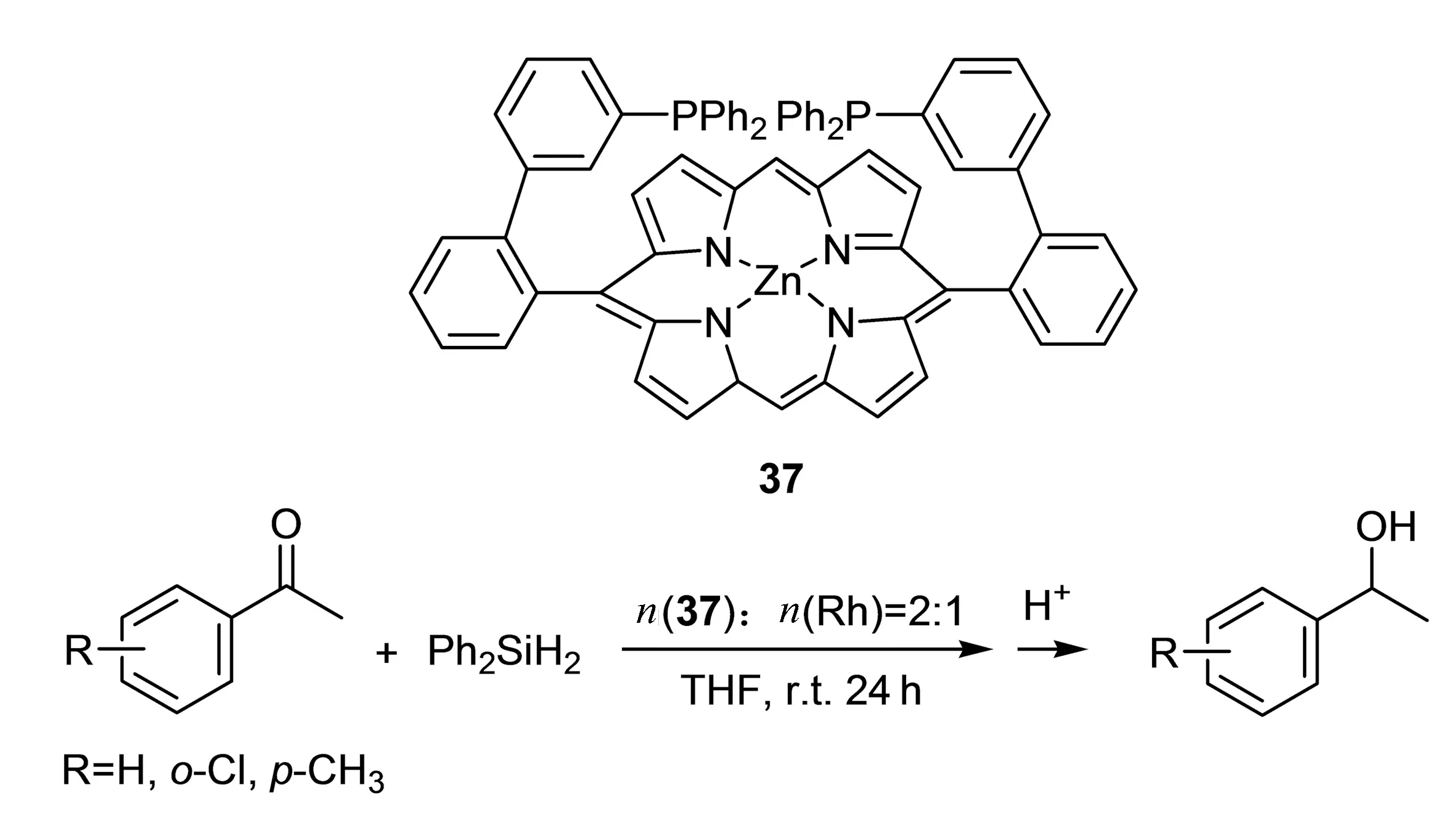
图21 [RhCl(COD)]2/37催化酮的硅氢加成反应Fig. 21 Hydrosilylation catalyzed with [RhCl(C2H4)2]2/37
Li课题组[39]报道了一系列含有炔基的铂-膦配合物催化体系(图22),与Speier催化剂、Karstedt催化剂进行比较,同样的催化条件下其催化的直链烯烃、苯乙烯同系物的硅氢加成反应具有较高的转化率和β-加成产物选择性.当用trans-Pt(PPh3)2(C≡CSiPh3)2来催化2-甲基苯乙烯、3-甲基苯乙烯、4-甲基苯乙烯、4-氟苯乙烯与三乙氧基硅烷的硅氢加成反应,在90 ℃下反应5 h时的转化率分别为92.4%,91.4%,89.4%,92.1%,β-加成产物选择性均在94%以上.相同条件下,催化1-己烯、1-十二烯与三乙基硅烷的硅氢加成反应,转化率均在91%以上,β-加成产物选择性均在97%以上.在有含硫化合物存在下,这种含炔基的铂-膦配合物催化体系也不失活,甲基硅烷基的存在对该催化剂催化硅氢加成反应过程有一定的影响.

图22 铂-膦配合物催化剂的合成Fig. 22 Synthesis of [Pt]/P complex catalyst
2005年,Hamze等[40]报道了氧化铂是催化芳基炔烃硅氢加成反应的一种很好的催化剂,但是由于无膦配体,其催化端炔与H—Si键加成反应时的反应选择性不高.2008年,Hamze等[41]合成了配体38,促进二氯化铂催化端炔与三乙基氢硅烷的硅氢加成反应(图23).当用苯乙炔、对甲基苯乙炔、对甲氧基苯乙炔、对溴苯乙炔等作为反应底物时,反应转化率90%以上,β-加成产物接近100%.
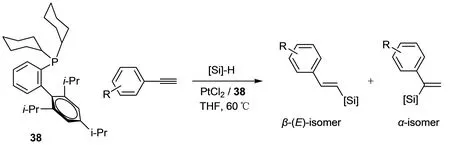
图 23 PtCl2/38催化炔烃的硅氢加成反应Fig. 23 Hydrosilylation of alkyne catalyzed with PtCl2/38

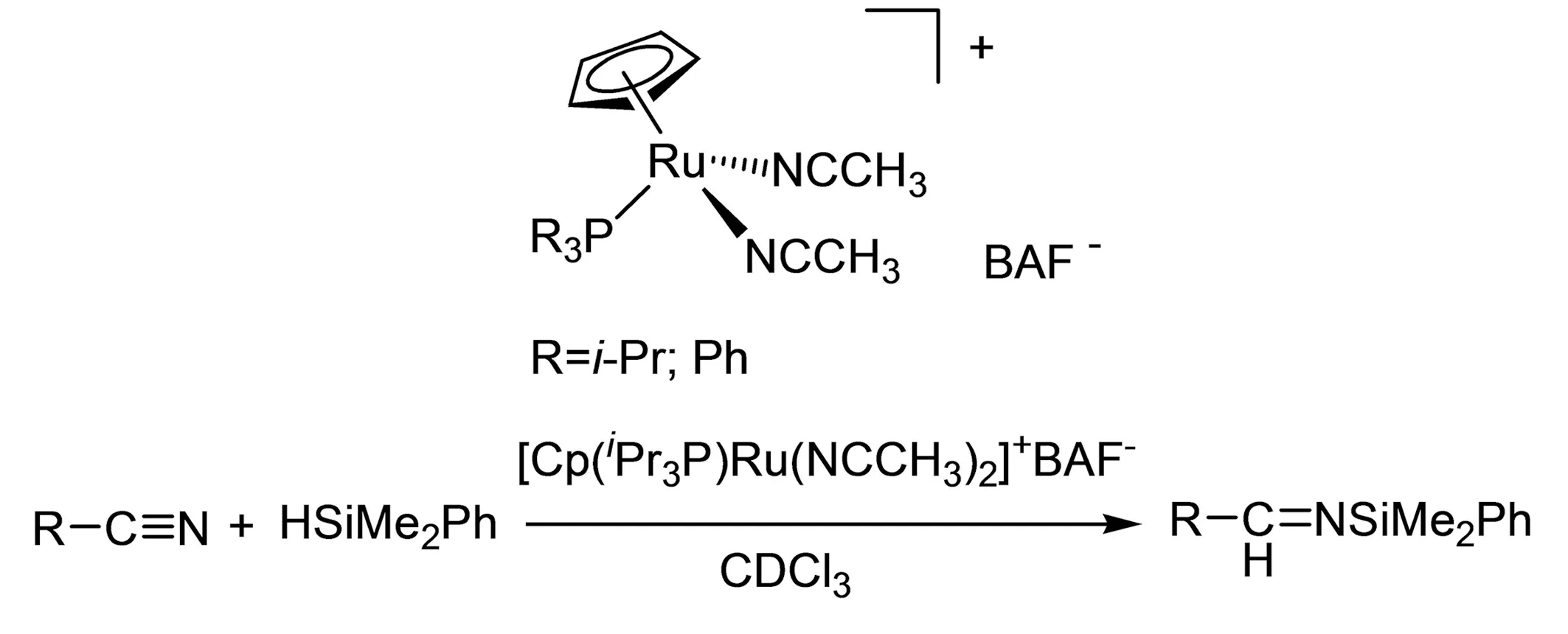
图24 [Ru]/P催化硅氢加成反应Fig. 24 Hydrogenation catalyzed with [Ru]/P
2017年,Wang等[44]合成了膦配体39,40,其与Co(acac)2形成配合物催化烯烃和苯基硅烷的硅氢加成反应(图25).常温下Co(acac)2/40催化体系催化苯乙烯和PhSiH3的硅氢加成反应的产率达98%以上,β-加成产物选择性为97%.而Co(acac)2/40配合物催化苯乙烯和Ph2SiH2的硅氢加成反应的支链和直链产物比则为36∶64.当用39作为配体且反应温度为50 ℃时,苯乙烯和PhSiH3的转化率达98%以上,直链产物占比为98%.将39,40配体与Co(acac)2用在催化1-辛烯、取代苯乙烯和PhSiH3、Ph2SiH2的硅氢加成反应中,均得到较好的反应结果.
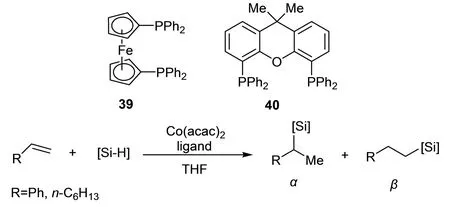
图25 Co(acac)2/39催化硅氢加成反应Fig. 25 Hydrogenation catalyzed with Co(acac)2/39
2 结论与展望
膦配体作为过渡金属催化反应中研究和应用最广泛的配体,在诸多催化反应中起着重要作用,可以提高反应转化率和目标产物选择性.通过调控膦配体的空间效应和电子效应,大量新颖膦配体被设计合成出来并用到过渡金属催化反应过程中.同时,含膦配体的过渡金属催化剂的循环利用、催化反应机理以及高效新颖的膦配体的合成等方面仍是未来研究的重点方向.
[1] MCGUINNESS D S, WASSERSCHEID P, KEIM W, et al. First Cr(III)-SNS complexes and their use as highly efficient catalysts for the trimerization of ethylene to 1-hexene[J]. J Am Chem Soc,2003,125(18):5272-5273.
[2] BOLLMANN A, BLANN K, DIXON J T, et al. Ethylene tetramerization: a new route to produce 1-octene in exceptionally high selectivities[J]. J Am Chem Soc,2004,126(45):14712-14713.
[3] DIJKSTRA H P, MEIJER M D, PATEL J, et al. Design and performance of rigid nanosize multimetallic cartwheel pincer compounds as Lewis-acid catalysts[J]. Organometallics,2001,20(14):3159-3168.
[4] DIJKSTRA H P, ALBRECHT M, VAN KOTEN G. Transcyclometalation, a versatile methodology for multiple metal-carbon bond formation with multisite ligands[J]. Chem Commun,2002,2(2):126-127.
[5] DE ARAUJO M P, DE FIGUEIREDO A T, BOGADO A L, et al. Ruthenium phosphine/diimine complexes: syntheses, characterization, reactivity with carbon monoxide, and catalytic hydrogenation of ketones[J]. Organometallics,2005,24(25):6159-6168.
[6] KOMURO T, ARAI T, KIKUCHI K, et al. Synthesis of ruthenium complexes with a nonspectator Si,O,P-chelate ligand: interconversion between a hydrido(η2-silane) complex and a silyl complex leading to catalytic alkene hydrogenation[J]. Organometallics,2015,34(7):1211-1217.
[7] RODRIGUES C, DELOLO F G, FERREIRA L M, et al. Ruthenium(III)/phosphine/pyridine complexes applied in the hydrogenation reactions of polar and apolar double bonds[J]. J Mol Struct,2016,1111:84-89.
[8] JESSOP P G, HSIAO Y, IKARIYA T, et al. Catalytic production of dimethylformamide from supercritical carbon dioxide[J]. J Am Chem Soc,1994,116(19):8851-8852.
[9] LI Y H, FANG X J, JUNGE K, et al. A general catalytic methylation of amines using carbon dioxide[J]. Angew Chem Int Ed,2013,52(36):9568-9571.
[10] DABBAWALA A A, SUDHEESH N, BAJAJ H C. Ru catalyzed formylation of diethylamine with CO2and H2under moderate pressure condition[J]. Indian J Chem,2015,54(6):752-756.
[11] BROWN C K, WILKINSON G J. Homogeneous hydroformylation of alkenes with hydridocarbonyltris-(triphenylphosphine) rhodium(I) as catalyst[J]. J Chem Soc A,1970,17(17):2753-2764.
[12] VAN DER SLOT S C, DURAN J, LUTEN J, et al. Rhodium-catalyzed hydroformylation and deuterioformylation with pyrrolyl-based phosphorus amidite ligands: influence of electronic ligand properties[J]. Organometallics,2002,21(19):3873-3883.
[13] YU S C, CHIE Y M, GUAN Z H, et al. Highly regioselective isomerization-hydroformylation of internal olefins to linear aldehyde using Rh complexes with tetraphosphorus ligands[J]. Org Lett,2008,10(16):3469-3472.
[14] TIJANI J, EL ALI B. Rhodium-catalyzed hydroformylation of olefins: effect of [bis(2,4-di-tert-butyl) pentaerythritol] diphosphite (alkanox P-24) on the regioselectivity of the reaction[J]. J Organomet Chem,2007,692(16):3492-3497.
[15] LABANDE A, DEYDIER E, MANOURY E, et al. Contribution of heterobifunctional ligands to transition metal-catalysed C—C coupling reactions[J]. Turk J Chem,2015,39(6):1158-1170.
[16] ACKERMANN L, KAPDI A R, SCHULZKE C. Air-stable secondary phosphine oxide or chloride (pre)ligands for cross-couplings of unactivated alkyl chlorides[J]. Org Lett,2010,12(10):2298-2301.
[17] NAGHIPOUR A, GHORBANI-CHOGHAMARANI A, BABAEE H, et al. Synthesis and X-ray structural characterization of a bidendate phosphine (dppe) palladium(II) complex and its application in stille and suzuki cross-coupling reactions[J]. Appl Organomet Chem,2016,30(12):998-1003.
[18] MORENO-MANAS M, PLEIXATS R, SERRA-MUNS A. Suzuki cross-couplings on aryl (heteroaryl) bromides and chlorides with bulky aliphatic phosphines/Pd(0)-triolefinic macrocyclic catalyst[J]. Synlett,2006(18):3001-3004.
[19] OGIWARA Y, MAEGAWA Y, SAKINO D, et al. Palladium-catalyzed coupling of benzoyl halides with aryltrifluorosilanes leading to diaryl ketones[J]. Chem Lett,2016,45(7):790-792.
[20] YANG J, LI P H, ZHANG Y C, et al. Dinuclear NHC-palladium complexes containing phosphine spacers: synthesis, X-ray structures and their catalytic activities towards the hiyama coupling reaction[J]. Dalton Trans,2014,43(19):7166-7175.
[21] NICOLAS E, OHLEIER A, ACCRISCIO F D, et al. "(Diphosphine)nickel"-catalyzed negishi cross-coupling: an experimental and theoretical study[J]. Chem Eur J,2015,21(21):7690-7694.
[22] ZHANG Z T, QIAO J F, WANG D, et al. Synthesis of isoflavones by room-temperature nickel-catalyzed cross-couplings of 3-iodo-(bromo)chromones with arylzincs[J]. Mol Divers,2014,18(2):245-251.
[23] PETUKER A, MERTEN C, APFEL U P. Modulating sonogashira cross-coupling reactivity in four-coordinate nickel complexes by using geometric control[J]. Eur J Inorg Chem,2015,2015(12):2139-2144.
[24] BERNHAMMER J C, CHONG N X, JOTHIBASU R, et al. Palladium(II) complexes bearing an indazole-derivedN-heterocyclic carbene and phosphine coligands as catalysts for the sonogashira coupling and the hydroamination of alkynes[J]. Organometallics,2014,33(13):3607-3617.
[25] KAUKORANTA P, KALLSTROM K, ANDERSSON P G. Microwave-assisted asymmetric intermolecular heck reaction using phosphine-thiazole ligands[J]. Adv Synth Catal,2007,349(17/18):2595-2602.
[26] MA M T, LU J M. Pd(II)-catalyzed oxidative heck-type reaction of triarylphosphines with alkenes via carbon-phosphorus bond cleavage[J]. Tetrahedron,2013,69(9):2102-2106.
[27] ACKERMANN L, BORN R, SPATZ J H, et al. Efficient aryl-(hetero)aryl coupling by activation of C—Cl and C—F bonds using nickel complexes of air-stable phosphine oxides[J]. Angew Chem Int Ed,2005,44(44):7216-7219.
[28] MARTIN R, BUCHWALD S L. Pd-catalyzed kumada-corriu cross-coupling reactions at low temperatures allow the use of knochel-type grignard reagents[J]. J Am Chem Soc,2007,129(13):3844-3845.
[29] BISCOE M R, BARDER T E, BUCHWALD S L. Electronic effects on the selectivity of Pd-catalyzed C—N bond-forming reactions using biarylphosphine ligands: the competitive roles of amine binding and acidity[J]. Angew Chem Int Ed,2007,46(38):7232-7235.
[30] ANDERSON K W, IKAWA T, TUNDEL R E, et al. The selective reaction of aryl halides with KOH: synthesis of phenols, aromatic ethers, and benzofurans[J]. J Am Chem Soc,2006,128(33):10694-10695.
[31] ZHANG H, LUO X C, WONGKHAN K, et al. Acceleration of reductive elimination of [Ar-Pd-Csp3]by a phosphine/electron-deficient olefin ligand: a kinetic investigation[J]. Chem Eur J,2009,15(15):3823-3829.
[32] MOTSWANINYANA W, ONANI M, LALANCETTE R, et al. Hemilabile imino-phosphine palladium(II)complexes: synthesis, molecular structure, and evaluation in heck reactions[J]. Chem Pap,2014,68(7):932-939.
[33] BEDFORD R B, DRAPER S M, SCULLY P N, et al. Palladium bis(phosphinite) ‘PCP’-pincer complexes and their application as catalysts in the suzuki reaction[J]. New J Chem,2000,24(10):745-747.
[34] KOSCHKER P, BREIT B. Branching out: rhodium-catalyzed allylation with alkynes and allenes[J]. Acc Chem Res,2016,49(8):1524-1536.
[35] LEE S Y, HARTWIG J F. Palladium-catalyzed, site-selective direct allylation of aryl C—H bonds by silver-mediated C—H activation: a synthetic and mechanistic investigation[J]. J Am Chem Soc,2016,138(46):15278-15284.
[36] NIYOMURA O, IWASAWA T, SAWADA N, et al. A bowl-shaped phosphine as a ligand in rhodium-catalyzed hydrosilylation: rate enhancement by a mono(phosphine) rhodium species[J]. Organometallics,2005,24(14):3468-3475.
[37] OCHIDA A, HAMASAKA G, YAMAUCHI Y, et al. Synthesis, properties, and catalytic applications of caged, compact trialkylphosphine 4-phenyl-1-phospha-4-silabicyclo[2.2.2]octane[J]. Organometallics,2008,27(21):5494-5503.
[38] SAITO M, NISHIBAYASHI Y, UEMURA S. Synthesis of dinuclear complexes bearing metalloporphyrin-phosphine hybrid ligands and their catalytic activity toward hydrosilylation of ketones[J]. Organometallics,2004,23(17):4012-4017.
[39] LI J Y, NIU C B, PENG J J, et al. Study on the anti-sulfur-poisoning characteristics of platinum-acetylide-phosphine complexes as catalysts for hydrosilylation reactions[J]. Appl Organomet Chem,2014,28(6):454-460.
[40] HAMZE A, PROVOT O, ALAMI M, et al. Platinum oxide catalyzed hydrosilylation of unsymmetrical internal aryl alkynes under ortho-substituent regiocontrol[J]. Org Lett,2005,7(25):5625-5628.
[41] HAMZE A, PROVOT O, BRION J D, et al. Platinum chloride/Xphos-catalyzed regioselective hydrosilylation of functionalized terminal arylalkynes[J]. Tetrahedron Lett,2008,49(15):2429-2431.
[42] GUTSULYAK D V, VYBOISHCHIKOV S F, NIKONOV G I. Cationic silane σ-complexes of ruthenium with relevance to catalysis[J]. J Am Chem Soc,2010,132(17):5950-5951.
[43] GUTSULYAK D V, NIKONOV G I. Chemoselective catalytic hydrosilylation of nitriles[J]. Angew Chem Int Ed,2010,49(41):7553-7556.
[44] WANG C, TEO W J, GE S Z. Cobalt-catalyzed regiodivergent hydrosilylation of vinylarenes and aliphatic alkenes: ligand- and silane-dependent regioselectivities[J]. ACS Catal,2017,7(1):855-863.
TheApplicationofTransitionMetal-phosphineLigandsinCatalyticReactions
YANG Chuang, LI Jiayun, PENG Jiajian, BAI Ying
(Key Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Hangzhou Normal University, Hangzhou 311121, China)
The phosphine ligands are the important ligands used in the homogeneous transition metal catalytic reaction. The steric and electronic effects of the phosphine ligands are especially important to the transition metal catalytic reaction. Therefore, in order to improve the conversion and selectivity of the catalytic reaction, the design and modification of the phosphine ligands are widely concerned. In this paper, the research progress of transition metal achiral phosphine complexes in the catalytic reactions is reviewed.
phosphine ligand; transition metal complexes; catalyst
2017-04-25
浙江省公益技术应用研究项目(2017C31105).
厉嘉云(1980-),女,高级实验师,博士,主要从事绿色催化研究.E-mail:jiayun1980@hznu.edu.cn;彭家建(1966-),男,研究员,博士,主要从事绿色催化研究.E-mail:jjpeng@hznu.edu.cn
10.3969/j.issn.1674-232X.2017.06.002
O621.2
A
1674-232X(2017)06-0567-13
猜你喜欢
中国饲料(2021年17期)2021-11-02
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
井冈山大学学报(自然科学版)(2021年1期)2021-03-05
工业催化(2020年9期)2020-11-13
化工时刊(2020年7期)2020-09-04
化工技术与开发(2020年8期)2020-08-26
无机化学学报(2020年7期)2020-07-20
心肺血管病杂志(2019年1期)2019-04-22
无机化学学报(2018年8期)2018-08-01
中国塑料(2015年8期)2015-10-14
