
Nonaka肌病临床病理及肌肉磁共振特点分析
2017-07-05 14:30周海涛任向阳马聪敏
中国实用神经疾病杂志 2017年10期
黄 超 周海涛 任向阳 马聪敏
郑州大学附属洛阳中心医院神经内科 洛阳 471000
Nonaka肌病临床病理及肌肉磁共振特点分析
黄 超 周海涛 任向阳 马聪敏
郑州大学附属洛阳中心医院神经内科 洛阳 471000
目的 探讨Nonaka肌病的临床、肌肉病理及肌肉磁共振特点。方法 入选2例患者,女性1例,男性1例,临床表现均以双下肢远端肌肉无力、萎缩为主,双上肢仅轻度受累。血清肌酸激酶轻度升高,肌电图提示肌源性损害,神经传导速度均正常。对患者完善大腿及小腿肌肉磁共振检查,并予以左上肢肱二头肌活检,进行组织学、酶组织化学及免疫组织化学染色,抽取外周静脉血2 mL送基因公司进行遗传性肌肉病相关基因测序。结果 肌肉病理提示,肌纤维肥大、萎缩、再生,肌纤维内可见镶边空泡,符合肌病样病理改变。肌肉MRI提示,大腿股四头肌脂肪化程度较轻,尤其是股外侧肌未受累及,大腿后组肌群及小腿胫前肌、胫后肌脂肪化程度严重。基因结果均提示GNE基因突变。结论 Nonaka肌病是一种与GNE基因突变相关的常染色体隐性遗传性远端肌病,临床表现特点为胫前肌首先受累,而股四头肌早期不受累。病理改变特点为肌纤维内镶边空泡形成。肌肉MRI可提示肌肉脂肪化的程度及分布规律,为诊断提供依据。
Nonaka肌病;GNE基因;肌肉磁共振
Nonaka肌病又称远端肌病伴镶边空泡(distal myopathy with rimmed vacuoles,DMRV)或遗传性包涵体肌病2型(hereditary inclusion body myopathy,hIBM2),是一种罕见的常染色体隐性遗传肌病[1],于1981年由日本学者Nonaka首先总结描述[2]。其临床特点为青壮年发病,以下肢胫前肌无力萎缩为主,早期股四头肌不受累。病理改变特点为肌纤维内镶边空泡形成,该病主要的致病基因为UDP-N-乙酰葡糖胺-2-表位酶/N-乙酰甘露糖胺激酶基因(GNE)[3]。目前国内自2003年以来陆续有该病的病例报道,多为总结其临床、病理及基因改变特点,而肌肉磁共振为近几年肌肉病领域飞速发展的热点,关于该病肌肉磁共振特点的报道尚少,因此,本文总结了2例Nonaka肌病的临床、病理及肌肉磁共振改变特点,现报道如下。
1 资料与方法
1.1 临床资料 病例1:患者女性,36岁,以“行走困难,双脚尖抬不起3 a”为主诉就诊。患者于3 a前,无明显诱因逐渐出现行走困难,双侧脚尖抬不起,易摔跤及扭伤脚踝,症状进行性加重,双侧小腿逐渐变细。无晨轻暮重感,不伴肢体麻木,不伴双上肢力弱。既往体健。父母非近亲结婚,家族成员中无类似疾病史。
神经系统检查:神志清,言语流利,脑神经外观(-),颈屈4级,肩内收/外展5级,握力5级,伸肘5-级,屈肘5级,伸屈腕左侧5-级,右侧5级,屈髋左5-级,右5级,双下肢内收5-级,外展5级,伸膝5级,屈膝4-级,双侧足背伸1级,跖屈5级,双侧膝腱反射(++),双侧跟腱反射(+)。双侧胫前肌萎缩。四肢深浅感觉检查正常。病理征未引出。
辅助检查:心电图正常;血清肌酸激酶797 IU/L;肌电图提示肌源性损害;感觉及运动神经传导速度正常。
病例2:患者男性,35岁,以“行走费力,上楼困难2 a”为主诉就诊。患者于2 a前无明显诱因出现行走费力,上楼困难,双侧脚尖抬不起,无晨轻暮重现象。症状逐渐加重,走平路易跌倒,双侧小腿变细,无肢体麻木感,无双上肢无力感。既往体健,父母非近亲结婚,家族成员中无类似疾病史。
神经系统检查:神志清,言语流利,脑神经外观(-),颈屈4级,肩内收/外展5级,握力5级,伸屈肘5级,伸屈腕5级,屈髋4级,双下肢内收4-级,伸膝5级,屈膝4-级,双侧足背伸2级,跖屈5级,双侧膝腱反射(++),双侧跟腱反射(+)。双侧胫前肌萎缩。四肢深浅感觉检查正常。病理征未引出。
辅助检查:心电图正常;血清肌酸激酶704 IU/L;肌电图提示肌源性损害;感觉及运动神经传导速度正常。
1.2 方法
1.2.1 肌肉活检:经医院伦理委员会批准,患儿家属同意,并签署知情同意书。对2例患者分别进行左侧肱二头肌活检和病理检查。标本在异戊烷预冷后,在液氮中冰冻固定,冷冻切片,进行组织学、酶组织化学和免疫组织化学染色,包括苏木精-伊红(HE)、改良Gomori三色(MGT)、高碘酸Schiff反应(PAS)、油红“O”脂肪染色(ORO)、三磷酸腺苷酶(ATP酶)(PH4.4~10.6)、还原型辅酶I四氮唑还原酶(NADH-TR)、琥珀酸脱氢酶(SDH)、肌营养不良素-N/C/R(dystrophin-N、C、R)、α/β/γ-肌聚糖蛋白(α/β/γ-sarcoglycan)、奇异不良素(dysferlin)。
1.2.2 肌肉MRI检查及评分:2例患者均行双侧大小腿MRI检查。由2名影像科医师独立进行评分。T1WI显示肌肉脂肪化程度,应用0~4分等级评估:0分:正常;1分:点状高信号;2分:高信号病变体积<30%;3分:高信号病变体积30%~60%;4分:高信号病变体积>60%。T2压脂像显示肌肉水肿程度。1.2.3 基因检测:在知情同意下,抽取2例患者外周血各2 mL送基因公司,进行遗传性肌肉病相关基因测序。
2 结果
2.1 肌肉病理结果 2例均呈肌病样病理改变。主要病理表现为肌纤维肥大、萎缩、再生,肌纤维内可见镶边空泡,HE染色可见空泡周边嗜碱性颗粒,MGT染色可见空泡周边红染,未见破碎红纤维,ORO及PAS染色可见肌纤维内空泡不着色,ATP酶染色显示两型纤维呈棋盘格分布,NADH-TR/SDH染色可见肌纤维内空泡氧化酶缺乏,空泡肌纤维略深染,COX未见深染及阴性肌纤维,NSE染色可见萎缩及空泡肌纤维深染,dystrophin-N/C/R,α/β/γ-sarcoglycan,dysferlin染色均可见肌纤维膜阳性表达。见图1。

图1 HE染色×200,部分肌纤维内可见镶边空泡(病例1)
2.2 肌肉MRI结果 2例患者大腿肌肉磁共振T1WI均提示股四头肌脂肪化程度较轻,其中股外侧肌均未受累及,而股后侧肌群脂肪化程度严重(表1)。2例患者小腿肌肉MRI均提示胫前肌及胫后肌脂肪化程度严重,其他肌肉均有不同程度的脂肪化(表1)。T2压脂像显示未见肌肉水肿。见图2。
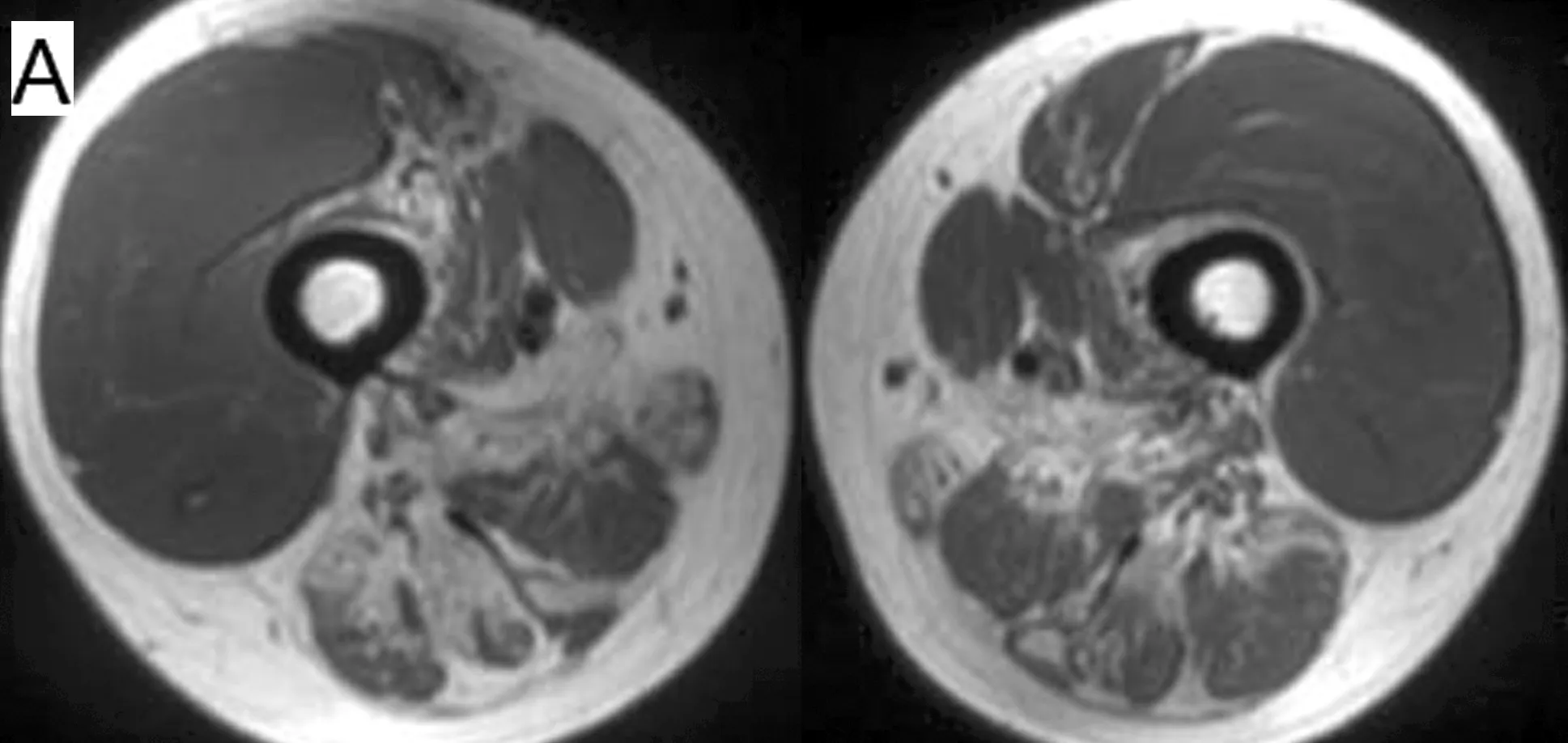

图2 病例2患者肌肉MRI T1WI序列 A:大腿中段肌肉MRI,提示股四头肌脂肪化程度较轻,股后侧肌群脂肪化程度较重;B:小腿中段肌肉MRI示,胫前肌、胫后肌脂肪化程度较重
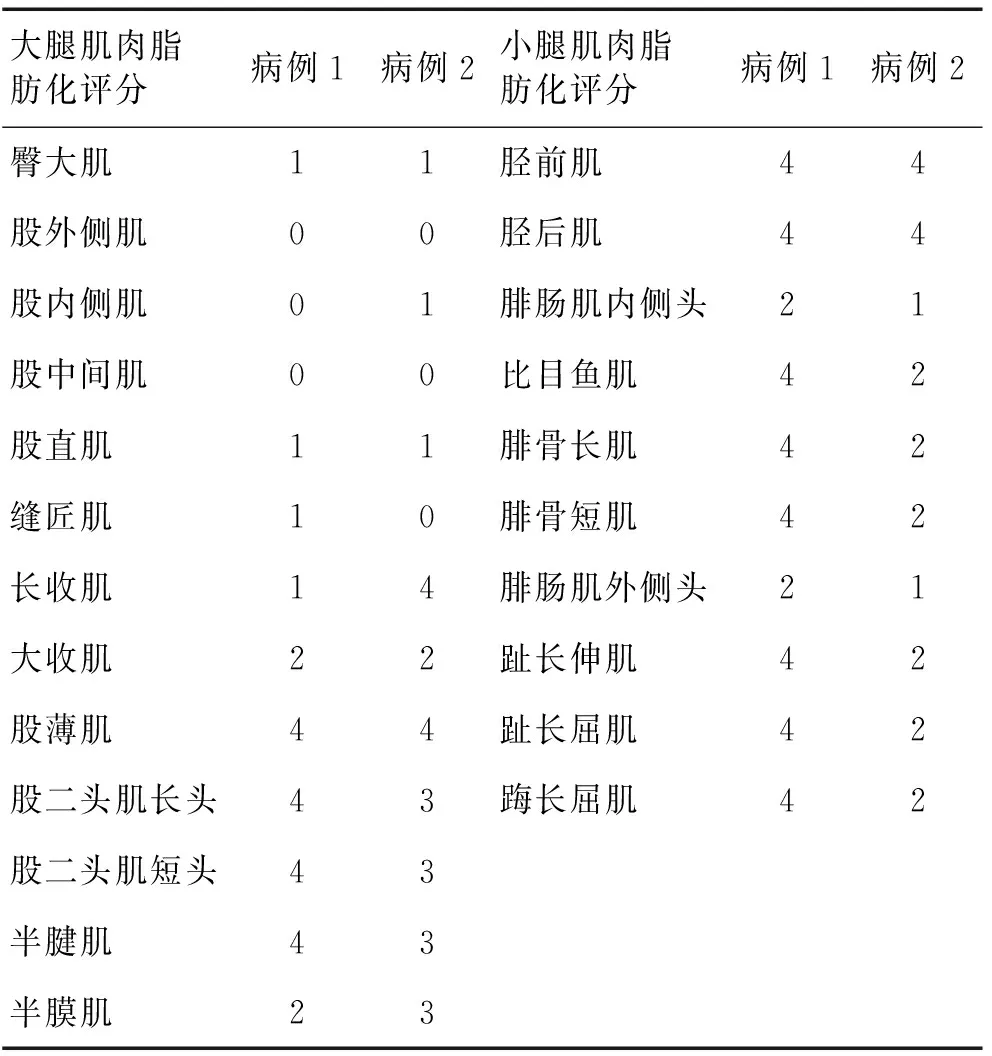
大腿肌肉脂肪化评分病例1病例2小腿肌肉脂肪化评分病例1病例2臀大肌11胫前肌44股外侧肌00胫后肌44股内侧肌01腓肠肌内侧头21股中间肌00比目鱼肌42股直肌11腓骨长肌42缝匠肌10腓骨短肌42长收肌14腓肠肌外侧头21大收肌22趾长伸肌42股薄肌44趾长屈肌42股二头肌长头43踇长屈肌42股二头肌短头43半腱肌43半膜肌23
2.3 基因分析结果 病例1 GNE基因的9号外显子存在c.1618C>T(p.H540Y)纯合突变。病例2 GNE基因的3号外显子存在c.527A>T(p.D176V)杂合突变及9号外显子存c.1571C>T(p.A524V)杂合突变。
3 讨论
本文2例均为青壮年患者,慢性起病,病情进行性发展,临床表现以双下肢远端无力为主,体检以双下肢足背伸力量差为主,伴胫前肌萎缩,无感觉障碍,血清肌酸激酶轻度升高,肌电图提示肌源性损害,神经传导速度正常,肌肉病理提示肌营养不良样病理改变伴镶边空泡形成,大小腿肌肉MRI提示股四头肌部分脂肪化程度轻微,胫前肌及胫后肌脂肪化严重,基因结果提示为GNE基因纯合或杂合突变,患者父母均正常,无类似疾病家族史,考虑遗传方式为常染色体隐性遗传。结合上述临床、病理、基因及影像学检查,2例患者均明确诊断为Nonaka肌病。
Nonaka肌病患者分布广泛,无人种之分,多于20~40岁发病,平均26岁,但也有10岁以下发病的报道。该病以双下肢远端肌肉无力为首发症状,可对称性或非对称性起病,典型特征为胫前肌首先受累,后逐渐累及腓肠肌,胸锁乳突肌,肢体近端肌肉亦可累及,疾病早期股四头肌不受累及,但发病5~10 a后部分患者会出现症状,有些患者甚至到疾病晚期股四头肌亦不受累及[4-5]。咽部及面部肌肉很少受累,仅见于少量文献报道[6]。该病可合并周围神经病变,特别是感觉神经受累,出现肢体的麻木感[7]。肌酸激酶为轻中度升高,肌电图提示肌源性损害`,部分患者可合并感觉神经传导传导速度及波幅下降[4]。本文2例患者发病年龄、起病方式及临床表现均比较典型,且无肢体麻木感,肌电图提示肌源性损害,神经传导速度正常,不合并周围神经病变。
Nonaka肌病主要的骨骼肌病理改变是RV形成。电镜下可见胞浆及细胞核内出现15~20 nm的管丝状包涵体,故也被称为包涵体肌病[3-4]。但RV并不是其特异性的病理改变,类似改变还可见于散发性包涵体肌炎,某些远端肌病及某些肌营养不良,如眼咽型肌营养不良等[8]。RV形成的机制尚不明确,推测可能与泛素降解和溶酶体降解途径的异常导致正常的蛋白胞质或胞核内异常聚集相关,亦有可能是肌核崩解的结果[3,8],而异常蛋白的沉积有可能诱发内质网应激和凋亡,从而导致肌纤维的损伤[9]。本文2例患者的病理表现较典型,但遗憾的是因条件所限,未能完善肌肉电镜下超微结构的检查。
GNE基因是该病已知的致病基因,其编码UDP-N-乙酰葡糖胺-2-表位酶/N-乙酰甘露糖胺激酶,该酶在唾液酸合成过程中催化前两步限速反应,故GNE基因突变引起肌细胞糖蛋白唾液酸化是Nonaka肌病重要的分子病理机制[10]。目前已知的突变位点广泛分布于编码表位酶及激酶区域,以错义突变为主,在世界多种族多国家均有报道,且部分种族有其热点突变,我国最常见的热点突变为P.L508S和P.D176V[11]。文中患者分别存在p.H540Y纯合突变及P.D176V、p.A524V杂合突变,其中P.D176V为热点突变,而其他突变在国内外Nonaka肌病患者中亦有散发报道,均为已知的突变位点。
肌肉MRI可判断骨骼肌受累肌群的分布及严重程度,明确肌病的分期及临床诊断,且对肌肉活检的取材部位具有指导意义,是近年来肌肉病领域发展的热点之一。正常骨骼肌MRI显示骨骼肌为长T1短T2信号,而脂肪化的肌肉显示短T1长T2的脂肪信号[12]。关于Nonaka肌病的骨骼肌MRI特点国内仅有个别报道,且样本量较少,均提示胫前肌及股后侧及内侧肌群受累较重,股四头肌受累较轻[11,13]。而国外有研究报道认为,不同时期受累肌群范围有所不同,疾病早期主要累及股二头肌,而疾病晚期股四头肌的股直肌、股内侧肌、股中间肌均可见脂肪浸润信号,而股外侧肌是唯一不受累的肌肉[14]。本文2例患者处于疾病早中期,MRI提示胫前肌、胫后肌及股后侧肌群受累较重,而股四头肌受累较轻,其中股外侧肌均未受累及,与国内外报道基本一致,但仍需累积大样本总结其变化发展规律。
Nonaka肌病是一种罕见的常染色体隐性遗传疾病,该病患者可予以唾液酸口服治疗[8,11],将大大减缓疾病的发展进程,甚至部分受累较轻的肌肉肌力可恢复正常,故早期识别该病,早期积极有效的治疗尤为重要。本文通过分析总结其临床、病理、基因及影像学特点,旨在提高大家对该病的认识,减少误诊、漏诊的概率,但样本例数太少,仍需国内大样本的总结研究。
[1] Huzing M,Krasnewich DM.Hereditary inclusion body myopathy:a decade of progress[J].Biochim Biophys Acta,2009,1 792(9):881-887.
[2] Nonaka I,Sunohara N,Ishiura S,et al.Familial distal myopathy with rimmed vacuole and lamellar(myeloid)body formation[J].J Neurol Sci,1981,51(1):141-155.
[3] 焉传祝,李大年,吴金玲,等.有镶边空泡的远端肌病九例临床和病理研究[J].中华神经科杂志,2003,36(2):138-139.
[4] 沈定国,吴士文.远端型肌病71例的临床及肌肉病理分析[J].中华神经科杂志,2005,38(4):222.
[5] Béhin A,Dubourg O,Laforêt P.Distal myopathy due to mutations of GNE gene:clinical spectrum and diagnosis[J].Rev Neurol(Paris),2008,164(5):434-443.
[6] 王朝霞,袁云,陈清棠.Nonaka肌病伴面部肌肉受累[J].中风与神经疾病杂志,2003,20:35-37.
[7] 丁卫江,曾国勇,邓丽影,等.有镶边空泡远端肌病合并周围神经病变一例[J].中华神经科杂志,2007,40(5):333-334.
[8] 李秀丽,陈琳,刘智.以镶边空泡为组织病理学特点的肌病研究进展[J].中华神经科杂志,2014,47(6):424-426.
[9] 陈涓涓,赵丹华,王朝霞,等.Nonaka肌病的骨骼肌存在内质网应激改变[J].中华神经科杂志,2012,45(1):11-13.
[10] 王朝霞,高云鹰,张英,等.Nonaka肌病的GNE基因突变研究[J].中风与神经疾病杂志,2006,23(2):201-203.
[11] 刘楠,张颖,苗晶,等.伴镶边空泡远端肌病患者GNE基因突变分析[J].中华神经科杂志,2015,48(1):32-34.
[12] Theodorou DJ,Theodorou SJ,Kakitsubata Y.Skeletal muscle disease:patterns of MRI appearances[J].Br J Radiol,2012,85(1 020):e1 298-1 308.
[13] 何杰,袁军辉.MRI在进行性肌营养不良中的应用价值[J].临床放射学杂志,2012,31(4):545-549.
[14] Tasca G,Ricci E,Monforte M,et al.Muscle imaging finding in GNE myopathy[J].J Neurol,2012,259(7): 1 358-1 365.
(收稿2016-10-21)
The clinicopathological characteristics and muscle MRI features of Nonaka myopathy
Huang Chao,Zhou Haitao,Ren Xiangyang,Ma Congmin
Department of Neurology,Luoyang Central Hosipital Affiliated to Zhengzhou University,Luoyang 471000,China
Objective To investigate the characteristics of both clinical pathology and muscle MRI in Nonaka myopathy.Methods The two included patients(one male and one female)who mainly represented weakness and atrophy in both lower limbs as well as slight abnormality in both upper limbs showed mild increase in creatine kinase(CK)level as well as myogenic abnormality and normal nerve conduction velocity observed in electromyogram(EMG).All patients
MRI in order to examine calf and thigh muscles and we used histological,enzyme-histochemical and immunohistochemical techniques to detect bicipital muscle of arm.Finally,peripheral venous blood of 2 mL was extracted to sequence related gene of hereditary muscular disorders,which was carried out by gene company.Results Muscle fibers presented by muscle biopsy showed atrophy,hypertrophy and regeneration and the presence of rimmed vacuoles(RV),which conformed to pathological changes of myopathy.Muscle MRI pointed out mild fatty infiltration in quadriceps,no affected changes in vastus lateralis muscle and severe fatty infiltration in posterior group of thigh muscles.The genetic analysis showed mutation of GNE gene.Conclusion Nonaka myopathy is a GNE gene-induced disease and inherited in an autosomal recessive manner,characterized by tibialis anterior muscles being affected initially and quadriceps being not affected at early stage.Typical muscle pathology shows the formation of rimmed vacuoles and the muscle MRI directly displays the degree and the distribution of muscle fatty infiltration.
Nonaka myopathy;GNE gene;Muscle MRI
R746
A
1673-5110(2017)10-0019-04
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26
中国临床医学影像杂志(2022年5期)2022-07-26
中国医院院长(2021年21期)2022-01-21
今日农业(2021年5期)2021-11-27
上海交通大学学报(2021年8期)2021-09-02
数字海洋与水下攻防(2021年2期)2021-05-08
国际放射医学核医学杂志(2021年10期)2021-02-28
医学信息(2020年12期)2020-07-27
舰船科学技术(2017年11期)2017-11-27
船海工程(2015年4期)2016-01-05
