
脊髓小脑共济失调3型发病机制研究进展
2017-04-05 05:22史长河许予明张化彪通讯作者
中国实用神经疾病杂志 2017年6期
张 帆 史长河 许予明 张化彪(通讯作者)
郑州大学第一附属医院 郑州 450002
脊髓小脑共济失调3型发病机制研究进展
张 帆 史长河 许予明 张化彪(通讯作者)
郑州大学第一附属医院 郑州 450002
共济失调;Ataxin-3;发病机制
脊髓小脑共济失调3型(Spinocerebellar ataxia type 3,SCA3)又称为马查德约瑟夫病(Machado-Joseph Disease,MJD),是我国遗传性共济失调(Hereditary Ataxia,HA)中最常见的亚型,约占所有遗传性共济失调的60%,其患病率为3~5/10万,仅我国就有4万余名患者[1-2]。该病以进展性小脑型共济失调为主要临床表现,主要包括步态不稳、肢体摇晃、动作准确性变差等,可伴眼外肌麻痹、吞咽困难、舌肌纤颤、锥体征及锥体外系征等其他临床表现[3-4],多数患者在起病后10~20 a内失去运动能力。目前此类疾病的常规治疗只能改善临床症状,缺乏有效治疗手段,给患者及其家庭造成极大的躯体、精神和经济负担,因此对脊髓小脑共济失调3型发病机制进行深入研究,寻找新的治疗靶点具有重要意义,本文就其致病基因、致病蛋白、包涵体形成、Ataxia-3毒性片段形成、蛋白质质量控制系统异常、转录异常、线粒体功能异常、RNA异常等方面做一简要综述。
1 SCA3的致病基因和致病蛋白Ataxin-3
1.1 致病基因—Ataxin-3基因 SCA3是一种常染色体显性遗传病,由Ataxin-3基因(又称为MJD1基因)突变所致。1994年,日本科学家Yoshiya Kawaguchi等[5]首先发现并报道了Ataxin-3基因。Ataxia-3基因位于14号染色体长臂,含有11个外显子,其10号外显子中有一段CAG重复序列(图1)[6]。正常人Ataxin-3基因CAG重复次数为12~44次,当CAG重复次数≥52次时,即会发病,而当CAG重复次数在45~51次时,疾病不完全外显(即可能会发病或发病时症状不典型)[7-8],因其由CAG重复过多而使其翻译形成过长的多聚谷氨酰胺链而致病,故SCA3是一种多聚谷氨酰胺病。
1.2 致病蛋白—Ataxin-3蛋白 Ataxia-3蛋白是一种广泛表达的去泛素化酶,分子量约42 kDa(千道尔顿),因多聚谷氨酰胺链长短不一,分子量会有所差异,该蛋白具有3个重要结构:Josephin结构域、泛素结合区(ubiquitin interacting motifs,UIM)和PolyQ序列(图1)。其Josephin 结构域具有去泛素化酶活性,UIM结构域则可特异地识别并结合泛素化的蛋白底物。Ataxia-3通过其UIM结构特异识别泛素化的蛋白底物,进而通过其Josephin结构域发挥去泛素化酶活性,使与其结合的底物去泛素化,调节底物蛋白的活性和稳定性[9-10]。Ataxia-3的PolyQ结构为其致病突变直接调控的结构,可抑制组蛋白乙酰化过程而调节转录过程。
2 发病机制
目前SCA3确切发病机制仍不十分清楚,下面将从包涵体形成、Ataxia-3毒性片段形成、转录异常、RNA异常、蛋白质质量控制系统异常、线粒体功能异常等方面进行叙述(图2)。
2.1 包涵体形成 神经元内广泛的包涵体形成是SCA3病人及动物模型最早发现的特征病理改变,主要位于细胞核内,此外轴突内亦可形成少量包涵体[11-12]。轴突内的包涵体可干扰神经元的轴浆运输,进而导致神经元功能受损;而核内包涵体形成的意义目前仍存在较大争议;有研究提示包涵体具有毒性作用,神经元内包涵体形成的多少与疾病严重程度成相关,包涵体中成分复杂,其中包括泛素、蛋白酶体组分、分子伴侣、转录因子、正常Ataxia-3蛋白等细胞内必需组分,这些重要物质聚集在包涵体内,不能正常发挥相应功能,进而影响细胞内多种代谢过程,导致细胞内环境紊乱[12-13];但也有学者认为包涵体形成具有保护作用,可将毒性蛋白聚集到包涵体中,避免对细胞的进一步损伤[11]。目前认为包涵体形成是由突变的Ataxia-3蛋白裂解而促发。
2.2 Ataxia-3毒性片段的形成 Ataxia-3蛋白可被钙蛋白酶(Calpain)裂解形成C端片段和N端片段,其中polyQ位于C端片段,正常情况下C端片段及N端片段均可被降解,不会聚集形成包涵体;突变的Ataxia-3被calpain裂解后,其含延长的polyQ的C-端片段形成不溶性的核内包涵体。体内动物实验发现抑制钙蛋白酶的活性可减少SCA3小鼠动物模型的包涵体形成,缓解其神经系统退行性变;增强钙蛋白酶的活性,则使SCA3小鼠模型的神经系统退行性变加重[14-15]。相应的体外实验显示增加钙蛋白酶的活性可促进Ataxia-3 C端毒性片段形成,使不溶性聚集体形成增加,并导致细胞凋亡增加[16]。且相对于全长的Ataxia-3蛋白裂解后形成含延长的polyQ的C端片段具有更强的毒性,故可将含延长polyQ的Ataxia-3的 C端片段视为毒性片段[14-16]。
2.3 转录异常 转录异常被认为在SCA3发病过程中起重要作用。在SCA3发病过程中,转录因子可被结合到核内包涵体中,使转录因子水平下调,不能发挥正常的转录调控功能,引起转录异常[17]。此外,突变的Ataxia-3失去对组蛋白乙酰化的抑制,组蛋白乙酰化水平升高,引起转录异常[18]。有研究显示,突变的Ataxia-3蛋白后,其亮氨酸拉链结构与DNA双链的GAGGAA富集区相互作用异常,影响转录[19-22]。Chou等[23]利用SCA3小鼠模型对脑组织进行转录组学研究,结果表明:谷氨酸能神经递质传递相关的基因、热休克蛋白(Heat Shock Protein,HSP)、调控神经细胞存活及分化的转录因子、伽马氨基丁酸受体亚基等的转录水平下调;介导神经细胞死亡的Bax、细胞周期蛋白D1等基因的转录水平上调。但SCA3疾病动物模型的转录情况和SCA3病人并不完全相同,转录异常的确切机制有待进一步研究。
2.4 RNA异常 在SCA3发病进展过程中,多种ataxia-3相关RNA存在异常。
Ataxia-3基因的CAG重复序列可编码含CUG重复序列的RNA。Li等[24]的研究表明,含过长CUG重复序列的RNA片段本身有毒性作用,在果蝇模型中,转录含过长CUG重复序列的RNA,可导致果蝇神经功能退化。进一步研究显示,含过长CUG重复序列的RNA可形成发夹样结构,并和MBNL1(Muscleblind Like 1)在细胞核中共定位,提示其可将MBNL1募集到细胞核中,使MBNL1介导的RNA可变性剪接发生异常,进而导致下游蛋白表达异常,并导致细胞功能异常[25]。
microRNA是一类内源性非编码的线性小RNA,通过与靶基因信使RNA(Manage RNA,mRNA)3’端非编码区特异性结合,调控靶基因表达。在SCA3病人血液中,发现miR-34b水平相比于正常人上调,miR-29a、miR25、miR-125b表达水平相比于正常人下调,提示miRNA参与SCA3发病过程[26]。此外,在SCA3动物模型中,抑制miRNA的表达,可加重动物模型的神经功能缺损和病例改变;在SCA3细胞系中,阻断miRNA可使Ataxia-3的毒性增加,并使细胞凋亡增加,提示干预miRNA可作为潜在靶点,但具体机制及干预方式仍需进一步研究[27]。
此外,非ATG起始的翻译过程、双向转录等使神经细胞产生多种异常的毒性产物(如聚丙氨酸、聚丝氨酸等),亦可能在SCA3发病中起到一定作用,影响神经细胞功能,但相关研究少,具体机制仍不明确[28]。
2.5 蛋白质质量控制系统异常 SCA3主要通过影响分子伴侣系统、内质网有关降解途径、泛素-蛋白酶体系统(ubiquitin-proteasome system,UPS)及自噬溶酶体通路(autophagy-lysosome pathway)等途径影响蛋白质降解过程。
热休克蛋白在调控蛋白质质量控制系统,介导蛋白质聚集及解聚,维持细胞内环境稳定等方面起重要作用[29]。在多种多聚谷氨酰胺病中,均发现在疾病早期热休克蛋白表达上调,增加错误折叠蛋白的在细胞中的溶解度,减少其聚集,而随着疾病进展,热休克蛋白的水平持续下调[30-31]。在SCA3病人脑组织中,发现HSP40、HSP90在包涵体中和Ataxia-3共定位;而在SCA3动物模型中,可见HSP40、HSP70的表达水平下调;此外,在SCA3病人来源的成纤维细胞中,发现HSP40的表达水平和SCA3的发病年龄相关联;而在SCA3细胞模型中过表达HSP40时,神经细胞内的包涵体形成减少,可见热休克蛋白在SCA3发病过程中起重要作用,上调热休克蛋白可作为潜在的治疗策略[23,32- 33]。
Ataxin 3参与调节内质网错误折叠蛋白的降解。VCP/p97蛋白(valosin-containing protein,VCP)可使内质网中错误折叠的分泌型蛋白离开内质网,并在蛋白酶体中降解,这一过程称为内质网有关降解途径(endoplasmic reticulum associated degradation,ERAD)[34]。Ataxia-3可与VCP/p97结合,形成Ataxin 3-VCP/p97复合体,参与ERAD过程,调控内质网错误折叠蛋白的降解[35]。突变的Ataxia-3蛋白可影响ERAD过程,突变的Ataxia-3与VCP/p97之间结合力增加,影响VCP/p97的底物蛋白从内质网中脱离,使VCP/p97的底物蛋白难以被转运至蛋白酶体降解,最终导致内质网错误折叠蛋白降解的异常,即ERAD异常[36]。
泛素-蛋白酶体系统的功能在SCA3中受损。对于突变的Ataxia-3,其去泛素化活性并无明显改变,但部分蛋白酶体组分被聚集在包涵体中,使蛋白酶体功能异常[37-38]。此外,泛素E3连接酶CHIP、Parkin在SCA3中表达下调,CHIP和Parkin均有神经保护作用。突变的Ataxia-3蛋白使Parkin的泛素化水平降低,促使Parkin通过细胞自噬途径降解增加,当Ataxia-3聚集形成包涵体时,可与Parkin结合使其进入包涵体中,导致细胞内Parkin量降低,Parkin底物降解异常[39- 40]。一些研究表明,突变的Ataxia-3与CHIP亲和力增加,在SCA3动物模型中,CHIP表达下调,使CHIP的底物蛋白降解异常,但其具体机制尚不明确[40-41]。
自噬溶酶体通路在降解大分子蛋白复合物及维持细胞内环境稳定的过程中起重要作用。一系列研究表明,自噬溶酶体通路在SCA3中受损[42-43]。Beclin1在自噬通路中起重要作用,在SCA3小鼠模型中,受累脑区Beclin1含量减少;过表达Beclin1,激活自噬系统,可缓解SCA3小鼠模型的神经功能缺损并减少神经元内包涵体形成,提示自噬异常在SCA3发病过程中起重要作用,调控自噬通路可作为潜在的治疗靶点[42-43]。
2.6 线粒体功能异常与细胞凋亡 既往研究表明,在亨廷顿病、脊髓小脑共济失调7型等多种polyQ疾病中,均存在线粒体能量代谢异常[44-45]。在SCA3细胞模型及动物模型中,亦发现细胞能量代谢障碍,其线粒体呼吸链复合体活性降低,导致自由基清除能力降低,自由基大量蓄积,一方面导致线粒体DNA损伤,DNA拷贝数降低,另一方面导致氧化压力增加,最终促发细胞凋亡[46-47]。在SCA3患者受累脑区进行病理染色,亦可见细胞凋亡的表现如核固缩、凋亡小体形成。目前关于线粒体功能异常及细胞凋亡在SCA3发病过程中所起到的作用仍不十分明确,有学者认为抗氧化治疗可能在SCA3治疗中起一定作用,但具体机制仍需要进一步研究。
3 总结与展望
SCA3目前发病机制尚未明确,现主要通过对症治疗提高患者的生活质量。而随着科学的发展,随着人类对这一疾病机制研究的不断深入,随着基因治疗、细胞治疗等新的治疗方式出现,必将为疾病治疗提供了新的思路,更有效的治疗靶点也会被不断发现。相信在不久的将来,人类对这一疾病定能找到更好的治疗办法。
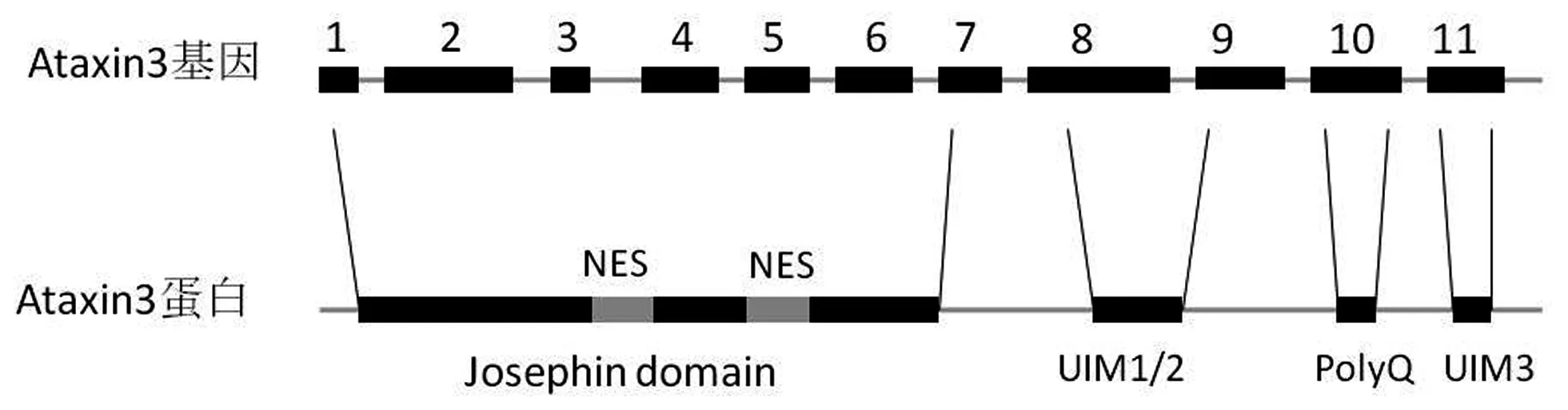
图1 Ataxin3基因及Ataxin3蛋白的基本结构
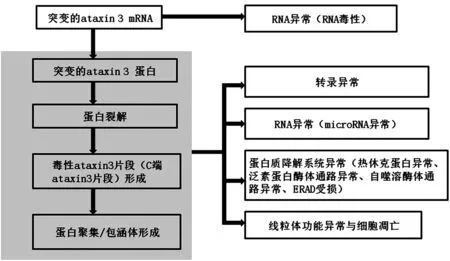
图2 SCA3发病机制模式图
[1] Gan SR,Shi SS,Wu JJ,et al.High frequency of Machado-Joseph disease identified in southeastern Chinese kindreds with spinocerebellar ataxia[J].BMC Med Genet,2010,11:47.
[2] Tang B,Liu C,Shen L,et al.Frequency of SCA1,SCA2,SCA3/MJD,SCA6,SCA7,and DRPLA CAG trinucleotide repeat expansion in patients with hereditary spinocerebellar ataxia from Chinese kindreds[J].Arch Neurol,2000,57(4):540-544.
[3] Riess O,Rub U,Pastore A,et al.SCA3:neurological features,pathogenesis and animal models[J].Cerebellum,2008,7(2):125-137.
[4] Rosenberg RN.Machado-Joseph disease:an autosomal dominant motor system degeneration[J].Mov Disord,1992,7(3):193-203.
[5] Kawaguchi Y,Okamoto T,Taniwaki M,et al.CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1[J].Nat Genet,1994,8(3):221-228.
[6] Heilig R,Eckenberg R,Petit JL,et al.The DNA sequence and analysis of human chromosome 14[J].Nature,2003,421(6 923):601-607.
[7] Evers MM,Toonen LJ,van Roon-Mom WM.Ataxin-3 Protein and RNA Toxicity in Spinocerebellar Ataxia Type 3:Current Insights and Emerging Therapeutic Strategies[J].Mol Neurobiol,2014,49(3):1 513-1 531.
[8] Padiath QS,Srivastava AK,Roy S,et al.Identification of a novel 45 repeat unstable allele associated with a disease phenotype at the MJD1/SCA3 locus[J].Am J Med Genet B Neuropsychiatr Genet,2005,133B(1):124-126.
[9] Burnett B,Li F,Pittman RN.The polyglutamine neurodegenerative protein Ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity[J].Hum Mol Genet,2003,12(23):3 195-3 205.
[10] Masino L,Musi V,Menon RP,et al.Domain architecture of the polyglutamine protein Ataxin-3:a globular domain followed by a flexible tail[J].FEBS Lett,2003,549(1-3):21-25.
[11] Seidel K,den Dunnen WFA,Schultz C,et al.Axonal inclusions in spinocerebellar ataxia type 3[J].Acta Neuropathologica,2010,120(4):449-460.
[12] Paulson HL,Perez MK,Trottier Y,et al.Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3[J].Neuron,1997,19(2):333-344.
[13] Chai Y,Wu L,Griffin JD,et al.The role of protein composition in specifying nuclear inclusion formation in polyglutamine disease[J].J Biol Chem,2001,276(48):44 889-44 897.
[14] Simoes AT,Goncalves N,Koeppen A,et al.Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant Ataxin 3 proteolysis,nuclear localization and aggregation,relieving Machado-Joseph disease[J].Brain,2012,135(8):2 428-2 439.
[15] Hubener J,Weber JJ,Richter C,et al.Calpain-mediated Ataxin-3 cleavage in the molecular pathogenesis of spinocerebellar ataxia type 3(SCA3)[J].Hum Mol Genet.,2013,22(3):508-518.
[16] Koch P,Breuer P,Peitz M,et al.Excitation-induced Ataxin-3 aggregation in neurons from patients with Machado-Joseph disease[J].Nature,2011,480(7 378):543-546
[17] Perez MK,Paulson HL,Pendse SJ,et al.Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation[J].J Cell Biol,1998,143(6): 1 457-1 470.
[18] Evert B O,Araujo J,Vieira-Saecker AM,et al.Ataxin-3 Represses Transcription via Chromatin Binding,Interaction with Histone Deacetylase 3,and Histone Deacetylation[J].J Neurosci,2006,26(44):11 474-11 486.
[19] Li F,Macfarlan T,Pittman RN,et al.Ataxin-3 is a histone-binding protein with two independent transcriptional corepressor activities[J].J Biol Chem,2002, 277(47):45 004-45 012.
[20] Araujo J,Breuer P,Dieringer S,et al.FOXO4-dependent upregulation of superoxide dismutase-2 in response to oxidative stress is impaired in spinocerebellar ataxia type 3[J].Human Molecular Genetics,2011,20(15): 2 928-2 941.
[21] Wang G,Sawai N,Kotliarova S,et al.Ataxin-3,the MJD1 gene product,interacts with the two human homologs of yeast DNA repair protein RAD23,HHR23A and HHR23B[J].Hum Mol Genet,2000, 9(12):1 795-1 803.
[22] Landschulz WH,Johnson PF,Mcknight SL.The leucine zipper:a hypothetical structure common to a new class of DNA binding proteins[J].Science,1988,240(4860):1 759-1 764.
[23] Chou A,Yeh T,Ouyang P,et al.Polyglutamine-expanded Ataxin-3 causes cerebellar dysfunction of SCA3 transgenic mice by inducing transcriptional dysregulation[J].Neurobiol Dis,2008,31(1):89-101.
[24] Li L,Yu Z,Teng X,et al.RNA toxicity is a component of Ataxin-3 degeneration in Drosophila[J].Nature,2008,453(7198):1 107-1 111.
[25] Mykowska A,Sobczak K,Wojciechowska M,et al.CAG repeats mimic CUG repeats in the misregulation of alternative splicing[J].Nucleic Acids Research,2011,39(20):8 938-8 951.
[26] Shi Y,Huang F,Tang B,et al.MicroRNA profiling in the serums of SCA3/MJD patients[J].Int J Neurosci,2013,124(2):97-101.
[27] Bilen J,Liu N,Burnett BG,et al.MicroRNA pathways modulate polyglutamine-induced neurodegeneration[J].Mol Cell,2006,24(1):157-163.
[28] Zu T,Gibbens B,Doty NS,et al.Non-ATG-initiated translation directed by microsatellite expansions[J].Proc Natl Acad Sci U S A,2011,108(1):260-265.
[29] Kampinga HH,Bergink S.Heat shock proteins as potential targets for protective strategies in neurodegeneration[J].Lancet Neurol,2016,15(7):748-759.
[30] Muchowski PJ,Schaffar G,Sittler A,et al.Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils[J].Proc Natl Acad Sci U S A,2000,97(14):7 841-7 846.
[31] Huen NY,Chan HY.Dynamic regulation of molecular chaperone gene expression in polyglutamine disease[J].Biochem Biophys Res Commun,2005,334(4):1 074-1 084.
[32] Zijlstra MP,Rujano MA,Van Waarde MA,et al.Levels of DNAJB family members(HSP40)correlate with disease onset in patients with spinocerebellar ataxia type 3[J].Eur J Neurosci,2010,32(5):760-770.
[33] Ito N,Kamiguchi K,Nakanishi K,et al.A novel nuclear DnaJ protein,DNAJC8,can suppress the formation of spinocerebellar ataxia 3 polyglutamine aggregation in a J-domain independent manner[J].Biochem Biophys Res Commun,2016,474(4):626-633.
[34] Zhong X,Pittman RN.Ataxin-3 binds VCP/p97 and regulates retrotranslocation of ERAD substrates[J].Hum Mol Genet,2006,15(16):2 409-2 420.
[35] Wang Q,Li L,Ye Y.Regulation of retrotranslocation by p97-associated deubiquitinating enzyme Ataxin-3[J].J Cell Biol,2006,174(7):963-971.
[36] Wang Q,Li L,Ye Y.Regulation of retrotranslocation by p97-associated deubiquitinating enzyme Ataxin-3[J].J Cell Biol,2006,174(7):963-971.
[37] Chai Y,Koppenhafer SL,Shoesmith SJ,et al.Evidence for proteasome involvement in polyglutamine disease:localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro[J].Hum Mol Genet,1999,8(4):673-682.
[38] Winborn B J,Travis SM,Todi SV,et al.The deubiquitinating enzyme Ataxin-3,a polyglutamine disease protein,edits Lys63 linkages in mixed linkage ubiquitin chains[J].J Biol Chem,2008,283(39):26 436-26 443.
[39] Durcan TM,Kontogiannea M,Thorarinsdottir T,et al.The Machado-Joseph disease-associated mutant form of Ataxin-3 regulates parkin ubiquitination and stability[J].Human Molecular Genetics,2010,20(1):141-154.
[40] Durcan TM,Fon EA.Ataxin-3 and Its E3 Partners:Implications for Machado-Joseph Disease[J].Front Neurol,2013,4:46.
[41] Scaglione KM,Zavodszky E,Todi S V,et al.Ube2w and Ataxin-3 coordinately regulate the ubiquitin ligase CHIP[J].Mol Cell,2011,43(4):599-612.
[42] Nascimento-Ferreira I,Nobrega C,Vasconcelos-Ferreira A,et al.Beclin 1 mitigates motor and neuropathological deficits in genetic mouse models of Machado-Joseph disease[J].Brain,2013,136(7):2 173-2 188.
[43] Nascimento-Ferreira I,Santos-Ferreira T,Sousa-Ferreira L,et al.Overexpression of the autophagic beclin-1 protein clears mutant Ataxin-3 and alleviates Machado-Joseph disease[J].Brain,2011,134(5):1 400-1 415.
[44] Ajayi A,Yu X,Lindberg S,et al.Expanded Ataxin-7 cause toxicity by inducing ROS production from NADPH oxidase complexes in a stable inducible Spinocerebellar ataxia type 7(SCA7)model[J].BMC Neurosci,2012,13(1):86.
[45] Goswami A,Dikshit P,Mishra A,et al.Oxidative stress promotes mutant huntingtin aggregation and mutant huntingtin-dependent cell death by mimicking proteasomal malfunction[J].Biochem Biophys Res Commun,2006,342(1):184-190.
[46] Kazachkova N,Raposo M,Montiel R,et al.Patterns of Mitochondrial DNA Damage in Blood and Brain Tissues of a Transgenic Mouse Model of Machado-Joseph Disease[J].Neurodegener Dis,2013,11(4):206-214.
[47] Emerit J,Edeas M,Bricaire F.Neurodegenerative diseases and oxidative stress[J].Biomed Pharmacother,2004,58(1):39-46.
(收稿2016-11-14)
R744.7
A
1673-5110(2017)06-0124-05
猜你喜欢
生物化学与生物物理进展(2022年8期)2022-08-20
世界科学技术-中医药现代化(2021年7期)2021-11-04
心血管病学进展(2021年8期)2021-09-13
现代临床医学(2021年1期)2021-01-26
山东医药(2021年28期)2021-01-11
科学导报·学术(2020年69期)2020-06-21
科学咨询(2020年1期)2020-02-11
中国脑血管病杂志(2019年8期)2019-03-13
华东师范大学学报(自然科学版)(2018年2期)2018-05-14
科学中国人(2017年36期)2017-06-09
