
代谢工程改造Corynebacteriumglutamicum生产L-苹果酸
2017-01-09 02:39赵一李天明刘金雷王崇慧仪宏冯惠勇
食品与发酵工业 2016年12期
赵一,李天明,刘金雷,王崇慧,仪宏,冯惠勇
(河北科技大学 生物科学与工程学院,河北 石家庄,050000)
代谢工程改造Corynebacteriumglutamicum生产L-苹果酸
赵一,李天明,刘金雷,王崇慧,仪宏,冯惠勇*
(河北科技大学 生物科学与工程学院,河北 石家庄,050000)
以提高L-苹果酸的产量为目标,采用一次交换两次同源重组的方法,利用反向筛选标记,在敲除了丙酮酸醌氧化还原酶编码基因(pqo),丙酮酸脱氢酶编码基因(pdh)和乳酸脱氢酶编码基因(lldh)的C. glutamicumΔpqoΔpdhΔlldh(C. glutamicumΔPPL)基础上,无痕敲除了L-苹果酸积累支流代谢途径的2个关键酶基因:苹果酸醌氧化还原酶编码基因(mqo)和苹果酸酶编码基因(male),同时敲入了苹果酸分泌转运蛋白基因(transb),获得了产L-苹果酸的工程菌株;采用高效液相色谱法检测了工程菌株C. glutamicumΔPPLΔmqo::transbΔmale的发酵产物。实验结果表明:C. glutamicum ATCC 13032发酵后不积累L-苹果酸,而工程菌C. glutamicumΔPPLΔmqo::transbΔmale发酵48 h,积累了12.8 g/L的L-苹果酸,工程菌的糖酸转化率为33.18%,为利用C. glutamicum ATCC 13032发酵生产L-苹果酸提供了基础遗传资源。
谷氨酸棒状杆菌(Corynebacterium glutamicum);L-苹果酸;基因敲除;基因敲入
L-苹果酸是一种十分重要的有机酸, 在食品、医药等工业中有着广泛的应用。L-苹果酸可用作食品添加剂,具有抑菌防腐的作用,可延长香肠和果酱的保存期[1];在葡萄酒酿造中用于除去酒石酸盐[2];作为日化品添加剂,能够起到润肤和抗皱作用[3];医药行业中,在抗疲劳、治疗心脏病、保护肝脏等方面有明显的作用[4]。
L-苹果酸的生产主要有化学合成法、酶催化法和微生物法[5]。化学合成法是利用顺丁烯二酸为原料经过两个步骤合成L-苹果酸,此方法存在污染环境,技术复杂,产物不纯等缺点[6]。第二种是酶催化法,以富马酸盐为原料,利用微生物的延胡索酸酶催化生成L-苹果酸,但此方法成本较高,产业化应用受到限制[7-9]。目前工业上有多种微生物法能够发酵生产L-苹果酸,如利用黄曲霉(Aspergillus flavus ATCC13697)以葡萄糖为原料,一步发酵生产苹果酸[10-13];也可以2种微生物联合应用生产L-苹果酸,如先利用少根根霉菌(Rhizopusahirrizus)将糖质类原料发酵成富马酸,再用膜醭毕赤酵母(Pichiamembranaefaciens)将富马酸转化为L-苹果酸[14-16]。与化学合成法和酶催化法相比,微生物转化法效率更高、能耗更低,而且清洁环保。
Corynebacterium.glutamicum是经典食品性安全菌,而且近年来,C. glutamicum ATCC 13032全基因组测序的完成和C. glutamicum ATCC 13032的遗传操作系统的完善,都为利用代谢工程手段对其进行定向改造提供了有利条件,使得C. glutamicum ATCC 13032作为细胞工厂,越来越广泛地应用于化学品的生物制造[17-20],因此用C. glutamicum ATCC 13032发酵生产L-苹果酸具有一定的理论及应用价值。C. glutamicum ATCC 13032积累L-苹果酸的代谢途径如图1所示。
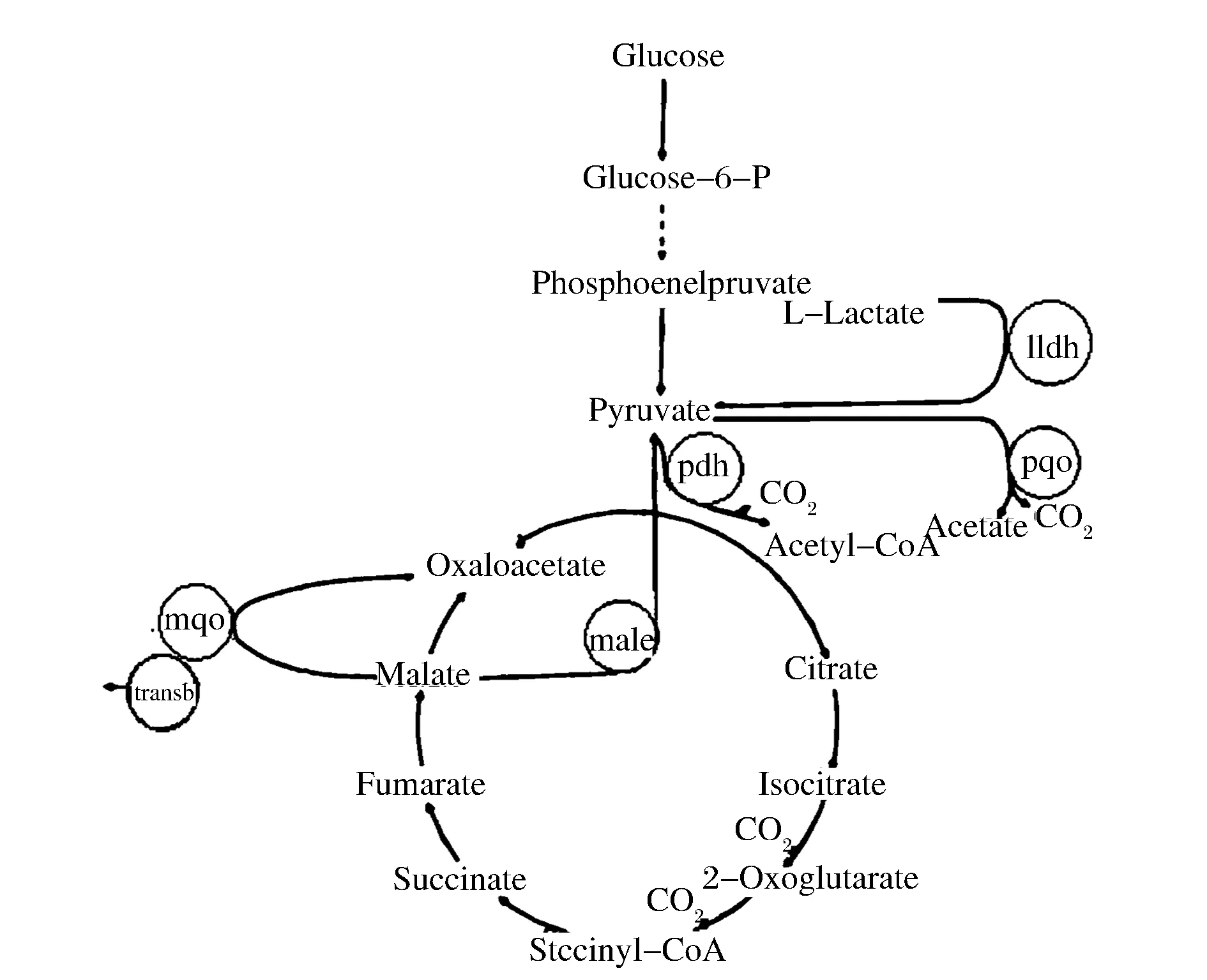
图1 C. glutamicum ATCC 13032部分代谢途径Fig.1 The partial metabolic pathways of C. glutamicum ATCC 13032
通过阻断乙酸和乳酸形成途径,提高丙酮酸的合成,以增加L-苹果酸合成的底物;通过阻断TCA循环途径中由L-苹果酸到草酰乙酸的代谢途径,大量积累L-苹果酸,使其接近1 mol葡萄糖生成1 molL-苹果酸的最大理论摩尔收率,获得具有工业应用价值、优良性能的工程菌。
本研究以实验室保存菌株C. glutamicum ΔPPL(实验室保存菌株)作为出发菌株,即敲除了丙酮酸醌氧化还原酶基因(pqo),丙酮酸脱氢酶基因(pdh)和乳酸脱氢酶基因(lldh)的工程菌,利用同源重组的原理及蔗糖致死基因(sacB基因)的反向筛选,无痕敲除了控制丙酮酸和L-苹果酸代谢过程中的支流代谢途径的关键基因:苹果酸酶编码基因(mqo)和苹果酸醌氧化还原酶编码基因(male),同时强化了L-苹果酸分泌转运蛋白基因(transb),获得了产L-苹果酸的工程菌株C. glutamicumΔPPLΔmqo::transbΔmale。
1 材料与方法
1.1 材料与试剂
1.1.1 菌株与质粒
本实验所用质粒与菌株见表1。

表1 本研究所用的菌株和质粒
1.1.2 试剂
High-Fidelity DNA 聚合酶购自北京全式金生物技术有限公司;限制性内切酶均购自NEB公司;T4DNA ligse购自大连宝生物工程公司;DNA分子量标准购自大连宝生物工程公司(TARAKA);基因组提取试剂盒、质粒小提中量试剂盒、超薄DNA产物纯化试剂盒和琼脂糖凝胶DNA回收试剂盒均购自天根生化科技(北京)有限公司;PCR引物由生工生物工程(上海)股份有限公司合成;其他试剂均为分析纯产品,购自生工生物工程(上海)股份有限公司。
1.1.3 培养基和菌株培养条件
LB培养基:10 g/L胰蛋白胨,5 g/L酵母提取物,10 g/L NaCl,调pH值6.5左右,需要时加入卡那霉素至终浓度30 mg/L。固体培养基则添加琼脂粉20 g/L。
CD培养基:1 g/LKH2PO4,3 g/L (NH4)2SO4,1 g/L NH4NO3,0.5 g/L MgSO4,0.2 g/L CaCl2,0.01 g/L Fe2(SO4)3,0.01 g/L MnSO4,0.002 g/L CuSO4,0.001 g/L ZnSO4,0.002 g/L生物素,pH值6.5左右。
转化培养基:CD培养基,25 g/L葡萄糖,5 g/L NaAce,pH值6.5左右。
种子培养基和发酵培养基:50 g/L葡萄糖,10 g/L NaAce,3 g/L (NH4)2SO4,1.5 g/L KH2PO4、0.5 g/L MgSO4·7H2O,5 g/L (NH4)2SO4,0.2 g/L CaCl2,pH 7.0。
C.glutamicumATCC 13032 在30 ℃、160 r/min条件下培养,E.coliDH5α在37 ℃、160 r/min条件下培养。
1.1.4 仪器与设备
PCR扩增仪,Eppendorf Mastercycler gradient;全自动凝胶成像系统,Bio-Rad Molecular Imager Gel DOC XR;台式高速离心机,Eppendorf Mini Spin;高速冷冻离心机,Thermo Sorvall Evolution RC;酶标仪,Bioteke Powerwave;高效液相色谱仪,Agilent 1260 Infinity。
1.2 方法
1.2.1 打靶质粒的构建
mqo基因敲除质粒构建方法为:分别以C.glutamicum ATCC 13032和酿酒酵母(Saccharomyces cerevisiae)的基因组作为模板,利用PCR扩增出待敲除基因mqo的上下游同源臂片段和待敲入的替换基因transb,利用融合PCR技术扩增获得目的片段mqoup-transb-do,纯化后与pK18mobsacB空质粒用Hind III和Not I进行双酶切反应,得到酶切产物,通过T4连接酶16 ℃过夜连接,并转化到E.coli DH5α感受态细胞中,通过验证获得打靶质粒pK18mobsacB/Δmqo::transb。同样,利用此方法构建获得male基因敲除质粒pK18mobsacB/Δmale,本实验所用引物如表2所示,下划线部分为酶切位点序列。

表2 本实验所用引物
1.2.2 感受态及电转化方法
大肠感受态制备参见《分子克隆实验指南》[21],谷氨酸棒状杆菌电转化感受态的制备参考《Handbook ofCorynebacteriumglutamicum》[22]。
1.2.3 工程菌株的转化
谷氨酸棒杆菌工程菌株的构建是利用负筛选标记通过2次同源重组的方法进行基因敲除来实现的,如图2所示。
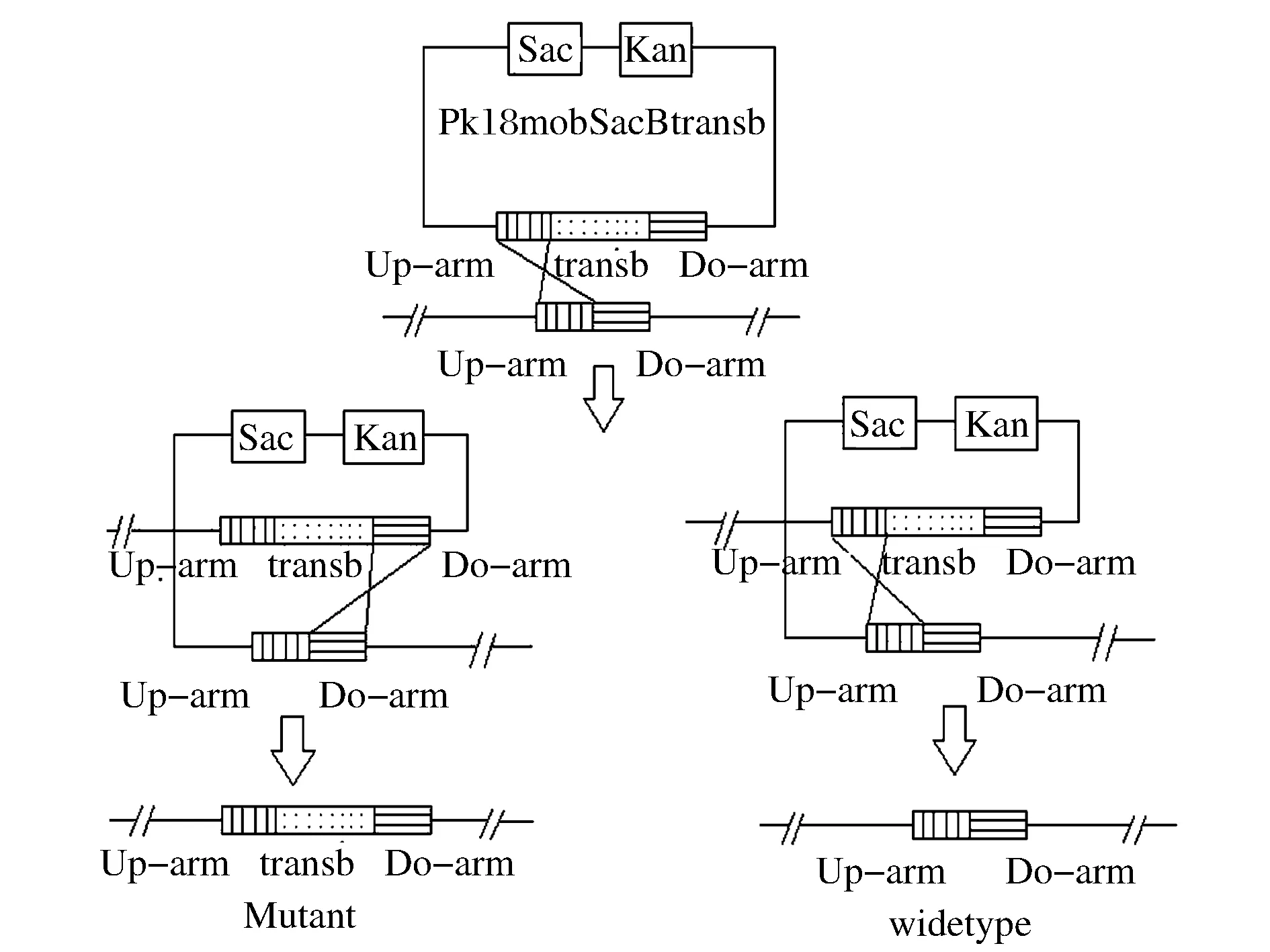
图2 同源双交换原理示意图Fig.2 Schematic diagram of homologous double switch
以获得突变菌株C.glutamicumΔPPLΔmqo::transb为例,首先将pK18mobsacB/Δmqo::transb经电转化(电击条件为,2.5 kV/cm电压,250 Ω电阻,25 μF电容,2 mm 电击杯)转入到工程菌C. glutamicumΔPPL感受态细胞中,经过电转的细胞再在含有0.5%乙酸钠的液体LB培养基中30 ℃恒温培养3 h,再把发酵液涂布在含有30 μg/mL卡那霉素及0.5%乙酸钠的LB固体培养基30 ℃恒温培养48~60 h。
1.2.4 阳性转化子的鉴定
经菌落PCR验证筛选出第1次同源重组的工程菌,即pK18mobsacB/Δmqo::transb经同源重组已整合到染色体上。再将其接到含有0.5%乙酸钠的液体LB培养基中,在30 ℃恒温摇床中培养12 h后,涂布在含有10%蔗糖的LB固体培养基上,利用蔗糖致死基因sacB基因负筛选出第2次同源重组的转化子,将其一一对应分别扩培到含10%蔗糖和30 μg/mL Kan抗性且含有0.5%乙酸钠的LB固体培养基中,培养1 d左右。最后挑选在蔗糖板上生长而Kan板上不生长的菌落,利用菌落PCR验证筛选出阳性工程菌C.glutamicumΔPPLΔmqo::transb。利用此方法将pK18mobsacB/Δmale转入C. glutamicumΔPPLΔmqo::transb中,获得C. glutamicumΔPPLΔmqo::transbΔmale。1.2.5 工程菌株生长及发酵L-苹果酸的研究
将验证正确的工程菌C. glutamicumΔPPLmqo::transb、C.glutamicumΔPPLmqo::transbΔmale和野生型菌株C. glutamicum ATCC 13032分别接种到含5%葡萄糖和1%乙酸钠的CD液体培养基中作种子,30 ℃、180 r/min过夜培养,培养物离心收集菌体,转移到以5%的葡萄糖1%乙酸钠作为碳源的CD培养基中,以相同条件继续摇床振荡培养,发酵48 h,整个发酵过程中定时调节pH,使之维持在7.0,同时测定OD值,定期记录菌株生长情况,并留存样品用来进行高效液相色谱的测定。
1.2.6 高效液相色谱检测丙酮酸和L-苹果酸的方法
将待测发酵液经转速10 000 r/min离心10 min,取上清液经0.22 μm 醋酸纤维素滤膜过滤后,利用高效液相色谱法分析L-苹果酸的含量[23]。检测条件为:Agilent1200高效液相色谱仪,紫外检测器,BDS HYPERSIL C18柱,4.6 mm×200 mm,流动相:乙腈和20 mmol/L KH2PO4(pH2.8)混合液(体积比为1∶99),流速0.5 mL/min。检测波长210 nm,柱温29 ℃,进样量20 μL。
2 结果与分析
2.1mqo基因缺失菌株的构建及验证
2.1.1mqo基因敲除打靶质粒的构建
苹果酸醌氧化还原酶基因(mqo)是以野生型C. glutamicum ATCC 13032基因组DNA为模板,经PCR扩增获得mqo基因的上下同源臂;再以Saccharomyces cerevisiae的基因组为模版,扩增出强化分泌的transb基因,融合获得大小为3 400 bp的片段mqoup-transb-do,如图3-A所示。将融合片段与空质粒pK18mobsacB相连,得到重组质粒pK18mobsacB/Δmqo::transb,对其进行双酶切验证,如图3-B所示,经酶切后的质粒分别获得大小为3 400 bp的融合片段和5 400 bp载体片段,符合与理论值一致,说明mqo基因敲除打靶质粒的构建成功。

A:1,2-mqo基因上下同源臂transb融合结果;M-DNA 5k marker; B:1-pK18mobsacB/Δmqo::transb双酶切;2-pK18mobsacB空质粒双酶切;M-DNA 15k marker图3 pK18mobsacB/Δmqo::transb打靶质粒的构建Fig.3 Construction of targeting plasmid pK18mobsacB/Δmqo::transb
2.1.2 工程菌株C. glutamicumΔPPLΔmqo::transb的筛选及验证
将重组质粒pK18mobsacB/Δmqo::transb电转化至实验室前期构建的工程菌C. glutamicumΔPPL感受态细胞中,通过两次同源重组使transb基因替换mqo基因,从而mqo基因被敲除。按照C. glutamicum ATCC 13032基因组序列中待敲除基因mqo的基因序列设计敲除验证引物QMQOF/QMQOR,随机挑取7株重组菌株,分别利用mqo基因敲除验证引物QMQOF/QMQOR、transb基因验证引物TRANSF/TRANSR进行菌落PCR验证,结果如图4所示,7号菌株扩增出1 000 bp的transb基因,而mqo基因未能扩增处,这说明7号菌株transb基因整合到了染色体,替换掉了mqo基因,从而获得了工程菌株C. glutamicumΔPPLΔmqo::transb。
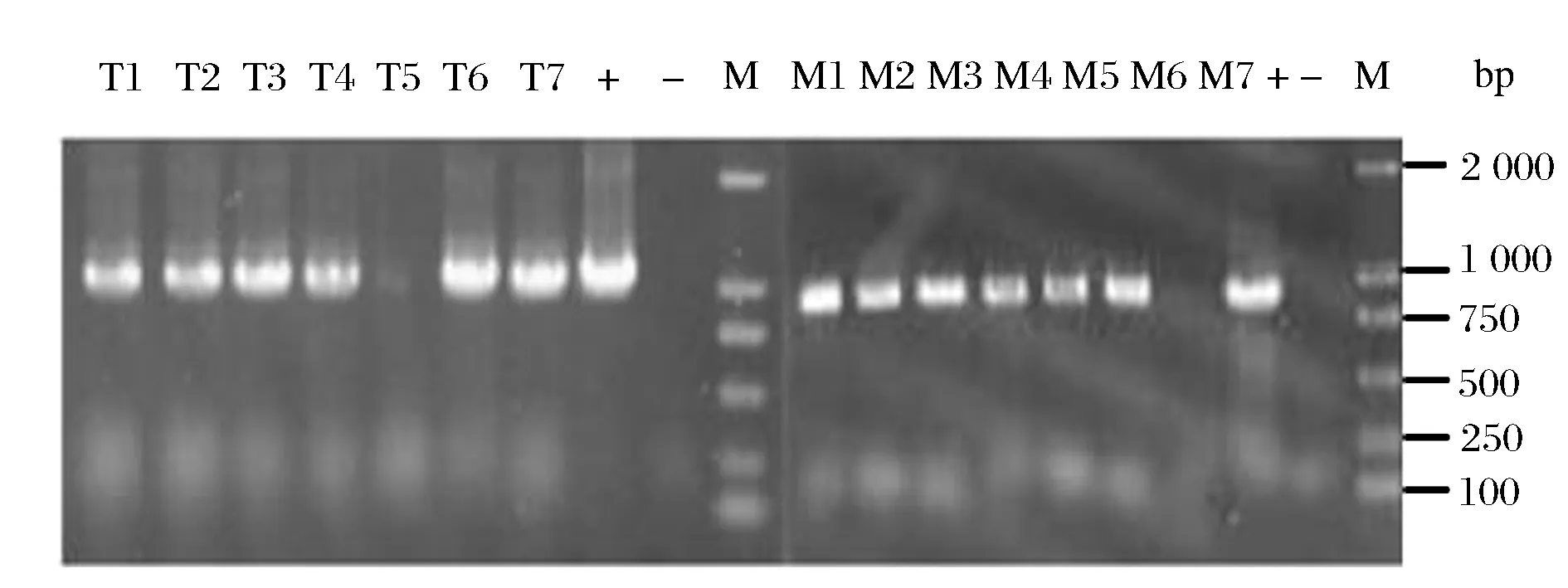
T1-7:transb基因验证结果;M1-7:mqo基因验证结果;+:阳性对照;-:阴性对照;M:DNA 2k marker图4 工程菌C. glutamicumΔPPLΔmqo::transb菌落PCR验证Fig.4 Colony PCR verification of C. glutamicum ΔPPL Δmqo::transb
2.2male基因缺失菌株的构建及验证
2.2.1male基因敲除打靶质粒的构建

A:1~3-male基因上下同源臂融合结果;M-DNA 2k marker;B:1-pK18mobsacB/Δmale双酶切;2-pK18mobsacB空质粒双酶切;M-DNA 15k marker图5 pK18mobsacB/Δmale打靶质粒的构建Fig.5 Construction of targeting plasmid pK18mobsacB/Δ mqo::transb
苹果酸酶基因(male)是以野生型C. glutamicum ATCC 13032基因组DNA为模板,经PCR扩增获得上下同源臂,经融合获得大小为1 900 bp的片段maleup-do,结果如图5-A。将融合片段与空质粒pK18mobsacB相连,得到重组质粒pK18mobsacB/Δmale,对其进行双酶切验证,如图5-B所示,经酶切后的质粒分别获得大小为1 900 bp的融合片段和5 400 bp载体片段,符合与理论值一致,说明male基因敲除打靶质粒的构建成功。2.2.2 工程菌株C. glutamicumΔPPLΔmqo::transbΔmale的构建
将重组质粒pK18mobsacB/Δmale电转化至工程菌C. glutamicumΔPPLΔmqo::transb的感受态细胞中,通过2次重组使male基因被敲除。按照按照C. glutamicum ATCC 13032基因组序列中待敲除基因male的基因序列设计敲除验证引物QMALEF/QMALER,随机挑取7株重组菌株,利用male基因敲除验证引物QMALEF/QMALER进行菌落PCR验证,结果如图6所示,2-5号菌株未能扩增出male基因,而阳性对照C. glutamicumΔPPLΔmqo::transb与其他菌株扩增出了male基因,阴性对照水为模版也同样未能扩增处条带,这说明2-5号菌株male基因被敲除,从而验证了工程菌株C. glutamicumΔPPLΔmqo::transbΔmale构建成功。

M1-10:male基因验证结果;+:阳性对照;-:阴性对照;M:DNA 2k marker图6 工程菌C.glutamicumΔPPLΔmqo::transbΔmale菌落验证 Fig.6 Colony PCR verification of C.glutamicumΔPPLΔmqo::transbΔmale
2.3 工程菌生长特性研究
将C.glutamicumΔPPL,C.glutamicumΔPPLΔmqo::transbΔmale与野生菌C. glutamicum ATCC 13032以等OD值分别接种到含5%葡萄糖和1%乙酸钠的CD培养基中,在30℃、160 r/min条件下培养,分别在12、24、36、48 h测定OD值,绘制生长状况图,如图7所示。由于基因缺失C. glutamicumΔPPL、C. glutamicumΔPPLΔmqo::transbΔmale的生长情况均不如野生型C. glutamicum ATCC 13032。与野生型C.glutamicum ATCC 13032相比,基因缺失菌株C.glutamicumΔPPL和C.glutamicumΔPPLΔmqo::transbΔmale生长较缓慢,而且其生长依赖于乙酸盐,是因为两株工程菌株都缺失了pdh基因造成的。
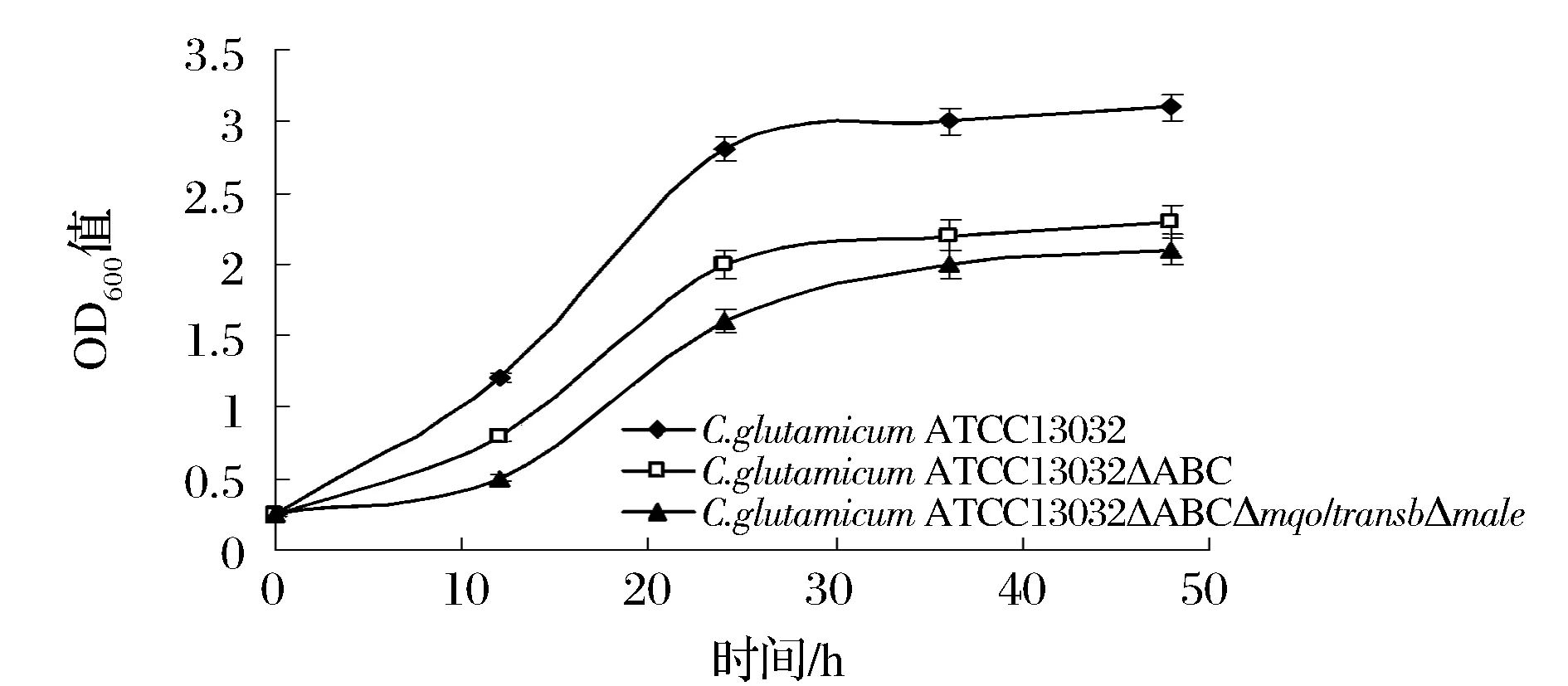
图7 生长曲线图Fig.7 Growth curve
2.4 工程菌的L-苹果酸产量
为了进一步验证工程菌L-苹果酸的产量,分别将C.glutamicumΔPPL、C. glutamicumΔPPLΔmqo::transbΔmale与野生菌C.glutamicum ATCC 13032同时以5%的葡萄糖和1%乙酸钠作为碳源的CD培养基进行摇瓶发酵,48 h后收集发酵液,利用高效液相色谱法测定发酵液中L-苹果酸的含量同时测定副产物丙酮酸的含量。由于工程菌 C. glutamicumΔPPL Δmqo::transbΔmale敲除了苹果酸代谢过程中的2个关键基因苹果酸醌氧化还原酶编码基因(mqo)和苹果酸酶编码基因(male),遏制了副产物的形成,且强化了苹果酸分泌基因(transb),因而积累的L-苹果酸明显高于野生型菌株和出发菌株。经计算结果表3所示。野生型菌株C. glutamicum ATCC 13032 发酵48小时后,没有L-苹果酸积累,丙酮酸的含量为0.65 g/L,而工程菌C.glutamicumΔPPL在敲除pqo,pdh, lldh后,丙酮酸含量提高为15.1 g/L、L-苹果酸含量为0 g/L,C. glutamicumΔPPLΔmqo::transbΔmale发酵48小时后L-苹果酸的含量为12.8 g/L、丙酮酸含量为14.6 g/L,L-苹果酸含量明显提升,丙酮酸含量略有下降,出峰结果如图8所示。
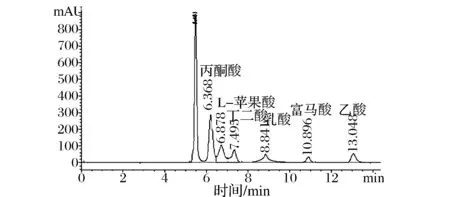
图8 工程菌C. glutamicum ΔPPLΔmqo::transb Δmale发酵48 h液相图Fig.8 C. glutamicumΔPPLΔmqo::transbΔmale 48 h fermentation phase diagram
3 结论
本文采用食品级微生物C.glutamicum作为L-苹果酸的生产菌,更安全可靠,符合食品添加剂的生产要求[24-25]。根据C.glutamicum的L-苹果酸生物合成的代谢途径,设计代谢工程改造的策略(如图1所示),在敲除了支流代谢相关酶编码基因基础上,依次构建了苹果酸转运蛋白酶编码基因(transb)置换苹果酸醌氧化还原酶编码基因(mqo)的打靶质粒pK18mobsacB/Δmqo::transb和敲除苹果酸酶基因(male)的打靶质粒pK18mobsacB/Δmale,通过电击转化,将打靶质粒pK18mobsacB/Δmqo::transb和pK18mobsacB/Δmale依次转化至已制备好的相应的感受态细胞中,通过一次交换两次同源重组的方法,经抗性筛选和反向标记筛选,获得阳性转化子,经PCR验证,证明C.glutamicumΔPPL染色体上的mqo,male基因被敲除,transb基因被敲入,获得C.glutamicumΔPPLΔmqo::transbΔmale工程菌株。在5%的葡萄糖1%乙酸为碳源的CD培养基中,进行摇瓶发酵,48 h后,野生菌株无L-苹果酸产生,而工程菌L-苹果酸的产量为12.8 g/L,工程菌的糖酸转化率为33.18%。有效地阻断了支流代谢途径,减少了副产物的生成,强化了L-苹果酸的分泌,提高了L-苹果酸的产率。获得的遗传资源具有产业化应用潜力,为进一步代谢工程改造菌种、提高L-苹果酸产量奠定了基础。
[2] 刘建军,姜鲁燕,赵祥颖,等.L-苹果酸的应用及研究进展[J].中国食品添加剂,2003(3):53-56.
[3] 汪多仁, 陈体庆.L-苹果酸的开发与应用[J]. 饮料工业, 2004, 7(1): 21-26.
[4] LIU G L, ZHOU Y, LUO H P, et al.A comparative evaluation of different types of microbial electrolysis desalination cells for malic acid production[J]. Bioresource Technology, 2015, 198: 87-93.
[5] KNUF C, NOOKAEW I, REMMERS I, et al. Physiological characterization of the high malic acid-producingAspergillusoryzaestrain 2103a-68[J]. Appl Microbiol Biotechnol, 2014,98(8): 3 517-3 527.
[6] 康毅,刘树文.苹果酸-乳酸发酵过程酒酒球菌碳源代谢分析研究进展[J].食品科学,2012,33(19):326-330.
[7] LITSANOV B, KABUS A, BROCKER M, et al. Efficient aerobic succinate production from glucose in minimal medium withCorynebacteriumglutamicum[J]. Microbial Biotechnology, 2012, 5(1): 116-128.
[8] NISHIMURA T, TERAMOTO H, INUI M, et al. Gene expression profiling ofCorynebacteriumglutamicumduring anaerobic nitrate respiration: induction of the SOS response for cell survival[J]. Journal of Bacteriology, 2011, 193(6): 1 327-1 333.
[9] 黄艳红, 田延军, 郝夕祥, 等.L-苹果酸代谢流分析及高产菌株构建[J]. 山东食品发酵, 2009(3): 3-8.
[10] 吴亚斌, 张梁, 石贵阳. 产L-苹果酸重组大肠杆菌的构建[J]. 生物加工过程, 2014(3): 12-18.
[11] 吴军林, 吴清平, 张菊梅.L-苹果酸的生理功能研究进展[J]. 食品科学, 2008,29(11): 692-695.
[12] LIU G L, LUO H P, WANG H H, et al.Malic acid production using a biological electrodialysis with bipolar membrane[J]. Journal of Membrane Science, 2014, 471: 179-184.
[13] 胡永红, 欧阳平凯, 沈树宝,等. 反应分离耦合技术生产L-苹果酸工艺过程的优化研究[J]. 生物工程学报, 2001, 17(5): 503-505.
[14] 周小燕, 吴清平, 蔡芷荷, 等. 曲霉N1-14′胞质酶活性与产L-苹果酸能力的关系[J]. 微生物学报, 2000, 30(5): 500-506.
[15] 田三德, 吴艳娜, 徐新丽. 混合发酵法生产L-苹果酸的工艺研究及浅析[J]. 食品科技, 2009, 30(3): 232-234.
[16] LITSANOV B, BROCKER M, BOTT M. Toward homosuccinate fermentation: metabolic engineering ofCorynebacteriumglutamicumfor anaerobic production of succinate from glucose and formate[J]. Applied and Environmental Microbiology, 2012, 78(9): 3 325-3 337.
[17] KIND S, JEONG W K, SCHRÖDER H, et al.Systems-wide metabolic pathway engineering inCorynebacteriumglutamicumfor bio-based production of diaminopentane[J]. Metabolic Engineering, 2010, 12(4): 341-351.
[18] ZAHOOR A, LINDNER S N, WENDISCH V F.Metabolic engineering ofCorynebacteriumglutamicumaimed at alternative carbon sources and new products[J]. Computational and Structural Biotechnology Journal, 2012, 3: 1-11.
[19] ZHU N, XIA H, WANG Z, et al.Engineering of Acetate Recycling and Citrate Synthase to Improve Aerobic Succinate Production inCorynebacteriumglutamicum[J]. PloS One, 2013, 8(4): e60659.
[20] SHI X C, CHEN Y, REN H F, et al. Economically enhanced succinic acid fermentation from cassava bagasse hydrolysate usingCorynebacteriumglutamicumimmobilized in porous polyurethane filler[J]. Bioresource Technology, 2014, 174: 190-197.
[21] EGGELING L,BOTT M.Handbook of Boca Raton[J].2005:535-545.
[22] HORTON R M.PCR-mediated.recombination and mutagenesis.SOEing together tailor-made gene[J].Molecular Biotechnology,1995,3(2):93-99.
[23] 尹志梅, 杨青, 吴敏秀,等.L-苹果酸定量测定方法[J]. 中国酿造,1999,18(4):33-34.
[24] KALINOWSKI J, BATHE B, BARTELS D, et al. The completeCorynebacteriumglutamicumATCC 13032 genome sequence and its impact on the production ofL-aspartate-derived amino acids and vitamins[J]. Journal of Biotechnology, 2003, 104(1): 5-25.
[25] MENTZ A, NESHAT A, PFEIFER-SANCAR K, et al. Comprehensive discovery and characterization of small RNAs inCorynebacteriumglutamicumATCC 13032[J]. BMC genomics, 2013, 14(1): 714.
Metabolic engineering ofCorynebacteriumglutamicumforL-malate production
ZHAO Yi, LE Tian-ming, LIU Jin-lei, WANG Chong-hui, YI YONG, FENG Hui-yong*
(College of Bioscience & Bioengineering,Hebei University of Science and Technology,Shijiazhuang 050000,China)
L-malate is an important organic acid in microbial metabolic pathway. It has been widely used in food, medicine and other industrial fields. To improve malate production, the C. glutamicum ΔpqoΔpdhΔlldh(C. glutamicum PPL) was genetically modified by homologous recombination technology and reverse selection method to knockout pyruvate quinone oxidoreductase (pqo), pyruvate dehydrogenase (pdh) andL-lactate dehydrogenase (lldh). The 2 key genes in malate metabolic pathway, malate quinone oxidoreductase (mqo) and malic enzyme (male), were knockout and one new gene malate transport-protein (transb) was inserted intoC.glutamicumΔPPL to obtain a new engineering mutantC.glutamicumΔPPL Δmqo::transbΔmale. TheL-malate production was measured by HPLC after fermentation for 48 h. The results showed that theL-malate production from fermentation broth of mutant was 12.8 g/L whereas the wild type had noL-malate accumulation. The sugar acid conversion rate of engineering bacteria was 33.18%. Our research provides the genetic resources for malate production byC.glutamicumfermentation.
Corynebacteriumglutamicum;L-malate;knockout;knockin
10.13995/j.cnki.11-1802/ts.201612003
硕士研究生(冯惠勇教授为通讯作者,E-mail:fenghuiyong@163.com)。
国家高技术研究发展计划(2014AA022102)
2016-05-20,改回日期:2016-07-18
猜你喜欢
牡丹江医学院学报(2021年5期)2021-12-05
江苏农业科学(2019年9期)2019-08-20
食品安全导刊(2018年30期)2019-01-28
湖北农业科学(2017年8期)2017-05-26
广东饲料(2016年1期)2016-12-01
分析仪器(2016年2期)2016-09-07
大陆桥视野·下(2016年6期)2016-08-06
中国果业信息(2013年12期)2013-01-22
中国烟草学报(2012年2期)2012-04-09
中国医学科学院学报(2012年3期)2012-03-25
