
CH2O→H2+CO小分子反应的理论研究
2015-12-22 11:03:19刘存海苏学军
海军航空大学学报 2015年5期
刘存海,苏学军,张 勇
(海军航空工程学院基础实验部,山东烟台264001)
CH2O→H2+CO小分子反应的理论研究
刘存海,苏学军,张 勇
(海军航空工程学院基础实验部,山东烟台264001)
利用量化计算的方法,在B3LYP/6-31G(d)水平上对CH2O→H2+CO小分子反应过程进行了研究,计算得到了反应物、过渡态及产物的分子构型。通过对分子结构参数的比较,对反应过程有了初步认识;通过理论计算得到了该反应的内禀反应坐标,从而确定了反应的具体路径,得到了该反应的活化能,并对反应物、过渡态及产物的电荷分布特点进行了分析。
甲醛;过渡态;虚频;活化能
甲醛,别名蚁醛,属于最简单的醛类物质,分子空间构型为平面三角形。它是一种无色气体,有强烈刺激性气味,熔点-118℃,沸点-19.5℃,易挥发,易溶于水、醇、醚等有机溶剂,通常以水溶液形式出现,35%~40%的甲醛水溶液称福尔马林。甲醛极易氧化,浓溶液长期放置,多个甲醛分子聚合成多聚甲醛。甲醛中毒表现为头晕、恶心等,严重的记忆力减退,甚至死亡。由于甲醛对人体健康的危害日益严重,人们对甲醛的各种性质和相关反应的研究越来越重视。李来才等[1]用密度泛函理论(DFT)的B3LYP方法,在6311++G(3df,3pd)基组水平上研究了CH2O与H自由基反应的微观机理,全参数优化了反应过程中各反应物、中间体、过渡态和产物的几何构型,在CCSD(T)水平上计算了它们的能量,振动分析结果证实了中间体和过渡态的真实性。翟巧玲等[2]用B3LYP,MP2和QCISD(T)方法并在6-311++G(3df,3pd)水平上计算了CH2自由基与O2在三重态势能面上的反应。通过内禀反应坐标(IRC)计算和振动分析,对反应过渡态进行确认,并确定了反应机理。黄多辉等[3]采用LSDA/6-311G(d,p)方法,研究了不同外电场对CH2O分子的基态键长、键角、总能量和电荷分布的影响。
过渡态理论又称活化络合物理论(Transition-Statetheory)或绝对反应速率理论,是1931-1935年由艾林(Eyring)和波兰尼(Polanyi)提出的,已被广泛应用于对化学反应过程的研究[4-9]。该理论的基本看法是当2个具有足够能量的反应物分子相互接近时,分子的价键要经过重排,能量要经过重新分配,方能变成产物分子,在此过程中要经过一过渡态,处于过渡态的反应系统成为活化络合物,这种状态极不稳定,可以分解为原始的反应物,也可以分解生成产物。本文运用Gaussian 09计算软件,利用过渡态理论对CH2O→H2+CO反应的分解过程进行了探讨,利用对内禀反应坐标IRC(Intrinsic Reaction Coordinate)计算的方法对反应路径进行了研究,得到了反应活化能,并通过电荷分析的方法,对反应物、过渡态及产物的电荷分布的特点进行了讨论。
1 计算方法
首先,猜测反应的中间过渡态的几何构型,并用GaussView构图软件构建了过渡态初始构型。利用Gaussian09计算程序,在B3LYP/6-31G(d)方法水平上对过渡态初始构型进行了结构优化和频率计算,得到了过渡态构型,频率分析确认计算所得构型即为CH2O→H2+CO反应的中间过渡态。然后,在同一方法水平上,对反应过程进行了IRC解析,从而得到反应物和产物的分子构型。其中,能量计算是在B3LYP/ 6-31++G(d)水平上进行的,零点能和频率计算分别采用0.961 3和0.892 9的修正因子进行校正[10-12]。
2 结果与讨论
2.1 反应过程解析
图1列出了优化得到的反应物Re、产物P、中间体IM和过渡态TS的几何构型。反应过程中,CH2O经过价键重排和能量的重新分配后转换为反应过渡态,由于过渡态具有不稳定性,随后过渡态再次经过价键和能量的改变生成最终的产物CO和H2,再由反应物到过渡态,以及过渡态到产物的过程中分子结构都要发生明显的变化,对应形成一系列的中间体。
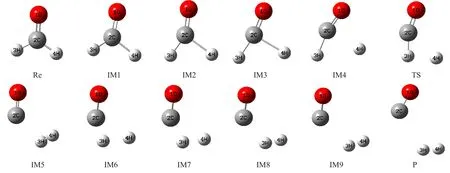
图1 反应过程中各驻点几何结构Fig.1 Geometries at the stationary points of the potential energy surface
表1列出了各种构型的键长、键角和二面角的具体数值。由于各个构型中的二面角∠4H-2C-1O-3H为0°或180°,可以确定各种构型中的原子均处在一个平面内。通过分析发现CH2O→H2+CO反应中CH2O分子中2个C-H键逐渐增大,2个H原子的距离逐渐缩小,由于2C-4H键的增大速率要大于2C-3H键的增大速率,致使2C-3H键首先断裂形成由CHO自由基和4H原子所构成的反应过渡态,即CH2O→H2+CO小分子反应的过渡态,然后2C-3H继续增大,最终断裂,3H原子与4H原子继续靠近,最终形成H2。
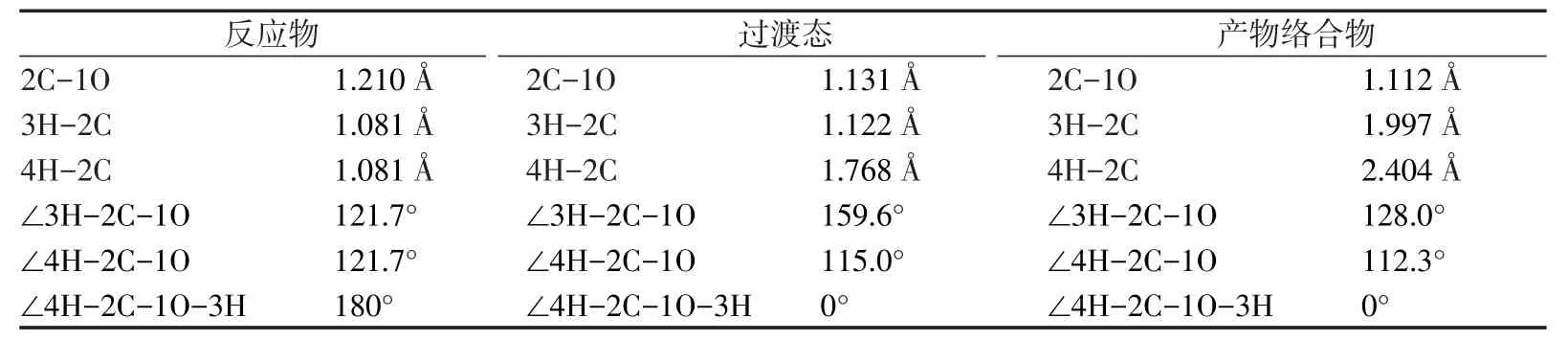
表1 反应物、产物和中间过渡态的结构参数Tab.1 Parameter of different structures of reactant,product and transition state
比较发现,C=O键的键长在反应过程中先增大后减小,主要是在由CH2O形成过渡态的过程中,2个H原子刚开始远离C原子时,2个H原子与C原子间的引力作用要大于O原子对C原子的作用力,致使O与C间的距离略有增大,而在由过渡态形成产物的过程中,由于2个H原子与C原子之间距离变大,H原子的作用力对C原子的作用力要小于O原子对C原子的作用力,故C=O键键长逐渐减小。图2为反应过渡态的振动模式,为2个C-H键的面内弯曲振动,与虚振动频率-2 205.18cm-1相对应。

图2 过渡态的振动模式Fig.2 Vibration mode of transition state
2.2 活化能计算
通过IRC解析,对CH2O→H2+CO小分子反应过程进行了研究,确定寻找到的反应过渡态能正确连接反应物CH2O和产物H2+CO,并得到了的反应的势能曲线,如图3所示。
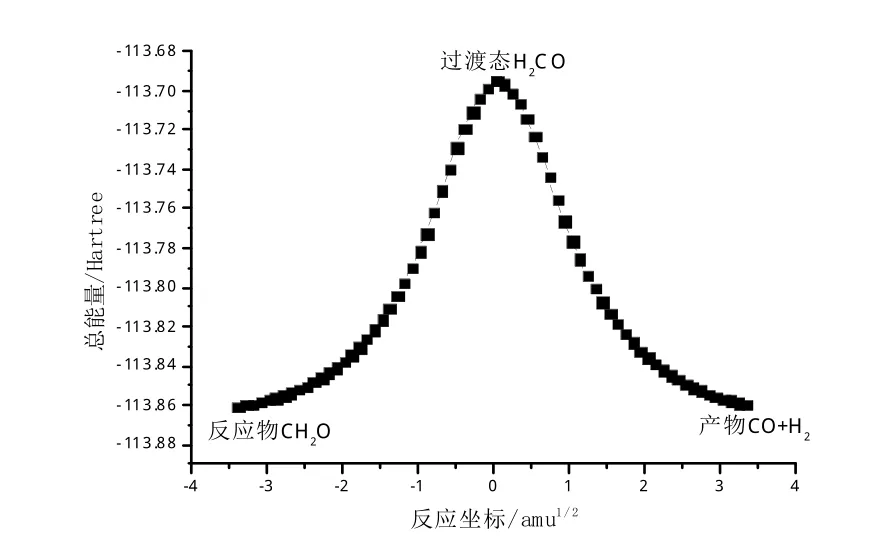
图3 反应势能曲线Fig.3 Curve of reaction potential energy
为进一步确定该小分子反应的特性,利用高精度能量计算的方法,得到CH2O→H2+CO小分子反应的正向、逆向反应活化能,见表2。不难发现,在CH2O→H2+CO的正向反应中须克服104.1kcal/mol的势垒高度,而在H2+CO→CH2O反应的逆过程中须克服103.4 kcal/mol的能量,由此可以确定H2+CO→CH2O反应的逆反应过程要比正反应的过程容易进行。

表2 反应物、过渡态和产物的能量及反应活化能Tab.2 Energy of reactant,product and transition state, as well as reaction activation
2.3 正电荷定域分布
为了进一步分析CH2O→H2+CO小分子反应的性质,对反应物、过渡态以产物及进行了Mulliken电荷分析。不同构型中,各原子的Mulliken电荷分布情况见图4。可以看出,反应物中电负性只存在于O原子上,而在过渡态和产物中,4H也呈现出电负性。此外,反应过程中O原子的所带负电荷逐渐增大,这主要是由于2个H原子对C=O结构的作用力逐渐减弱,致使O原子电负性增强。

图4 Mulliken电荷分布Fig.4 Mulliken charge distribution
3 总结
通过对CH2O→H2+CO小分子反应的研究,对反应过程中的分子结构变化特点有了初步了解。利用IRC解析对该反应的反应过程进行了确认,得到了反应的正向、逆向反应活化能分别为104.1kcal/mol和103.4kcal/mol,从而得出该反应的逆反应过程要比正反应的过程容易的进行的结论。此外,利用对反应前后反应物、过渡态和产物进行的Mulliken电荷分析,了解了反应过程中各原子上所带电荷的变化过程。
[1]李来才,朱元强.CH2O与H反应机理的量子化学研究[J].四川师范大学学报:自然科学版,2005,28(2):215-217. LI LAICAI,ZHU YUANQIANG.Quantum chemistry study on the reaction mechanism of CH2O and H[J].Journal of Sichuan Normal University:Natural Science,2005,28(2):215-217.(in Chinese)
[2]翟巧玲,黄浩,吕文阳,等.CH2自由基与O2反应机理的研究[J].西南师范大学学报:自然科学版,2006,31(1):92-94. ZHAI QIAOLING,HUANG HAO,LV WENYANG,et al.Study on the mechanism for the CH2+O2reaction[J]. Journal of Southwest China Normal University:Natural Science Edition,2006,31(1):92-94.(in Chinese)
[3]黄多辉,万琴,程晓洪.外电场作用下CH2O分子的结构与特性[J].宜宾学院学报,2010,10(12):54-56. HUANG DUOHUI,WAN QINB,CHENG XIAOHONG. Structure and characteristics of CH2O molecule under external electric field[J].Journal of Yibin University,2010,10(12):54-56.(in Chinese)
[4]宋朋,胡瑞,赵金峰.丁酸甲酯高温分解的过滤态理论研究[J].辽宁大学学报:自然科学版,2014,41(3):216-221. SONG PENG,HU RUI,ZHAO JINFENG.Transition state theory study on methyl butanoate decomposition reaction[J].Journal of Liaoning University:Natural Sciences Edition,2014,41(3):216-221.(in Chinese)
[5]WU SHILIANG,SHEN DEKUI,GAO SHANYUN. Thermodynamic analysis and transition state study for pyrolysis of levoglucosan and glyceraldehydes through quantum simulation[J].Journal of Southeast University:English Edition,2013,29(3):282-288.
[6]WANG ZUNJING,CHEN MIN,GUO ZENGYUANT. Analysis of transition state theory for condensation[J]. Chinese Science Bulletin,2002,47(11):952-954.
[7]SUN YOUMIN,WU JUNSEN,LIU CHENGBU.A comparison of transition state of phenol in H-atom abstraction by methyl and methylperoxyl radicals[J].Chinese Science Bulletin,2007,52(9):1182-1186.
[8]王成云,顾慰中.配合物氧化还原反应两种机理的过渡态[J].曲阜师范大学学报:自然科学版,2001,27(1):59-61.WANG CHENGYUN,GU WEIZHONG.Transition state of two sphere in oxidation-reduction of coordnation compound[J].Journal of Qufu Normal University:Natural Science,2001,27(1):59-61.(in Chinese)
[9]赵衍辉,刘杰,张新.孤立条件下α-丙氨酸分子碳骨架异构的过渡态理论研究[J].白城师范学院学报,2014,28(3):6-8. ZHAO YANHUI,LIU JIE,ZHANG XIN.Transition state theory study on the isomerism of alpha alanine carbon skeleton under isolated condition[J].Journal of Baicheng Normal University,2014,28(3):6-8.(in Chinese)
[10]LEE C,YANG W T,PARR R G.Development of the colle-salvetti correlation-energy formula into a functional of the electron density[J].Physical Review B,1988,37(2):785-789.
[11]BECKE D A.Density-functional exchange-energy approximation with correct asymptotic behavior[J].Physical ReviewA,1988,38(6):3098-3102.
[12]MIEHLICH B,SAVIN A,STOLL H,et al.Results obtained with the correlation energy density functionals of Becke and Lee,Yang and Parr[J].Chemical Physics Letters,1989,157(3):200-206.
Theoretical Research on the Reaction of Small Molecule CH2O→H2+CO
LIU Cunhai,SU Xuejun,ZHANG Yong
(Department of Basic Experiment,NAAU,Yantai Shandong 264001,China)
In this paper,the reaction of small molecule CH2O→H2+CO was researched at the level of B3LYP/6-31G(d),us⁃ing the method of quantum calculation,and the structures of reactant,product and transition state were gained.By compar⁃ing the parameter of different structures,the reaction process was generally known.At the same time,the specific reaction path and activation energy was determined by the theoretical calculation to the intrinsic reaction coordinate.At last,the distributing characters of charges in reactant,transition state and product was analyzed.
formaldehyde;transition state;imaginary frequency;activation energy
O604
A
1673-1522(2015)05-0497-04
10.7682/j.issn.1673-1522.2015.05.020
2015-07-08;
2015-08-03
刘存海(1980-),男,工程师,硕士。
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
大学化学(2021年8期)2021-09-26 10:51:16
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08 00:48:08
电脑知识与技术(2018年3期)2018-03-21 09:27:04
中学化学(2017年5期)2017-07-07 08:40:47
北京航空航天大学学报(2017年10期)2017-04-20 08:51:23
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
中学化学(2016年4期)2016-05-30 16:20:37
航天返回与遥感(2014年4期)2014-07-31 17:47:47
中学化学(2014年1期)2014-04-23 08:59:04
