
工业微生物基因组编辑技术的研究进展
2015-07-19 13:08朱林江李崎
生物工程学报 2015年3期
朱林江,李崎
工业微生物基因组编辑技术的研究进展
朱林江1,2,李崎1,2
1江南大学生物工程学院工业生物技术教育部重点实验室,江苏无锡 214122 2江南大学食品安全与营养协同创新中心,江苏无锡 214122

朱林江, 李崎. 工业微生物基因组编辑技术的研究进展. 生物工程学报, 2015, 31(3): 338–350.Zhu LJ, Li Q. Genome editing of industrial microorganism. Chin J Biotech, 2015, 31(3): 338–350.
近年来,基因组范围的高效编辑技术发展迅速,对工业微生物基因组的改造效率不断提升,彻底改变了以“一次操作、一个抗性基因、一个修饰位点”为特征的传统遗传操作模式,实现了基因组上多重位点的同步编辑,精确高效且无需抗生素辅助的插入替换或删除,以及大片段基因组DNA的剪切-粘贴等。这些技术的应用,能够高效构建优良性能的生产菌株,必将推动传统发酵产业的革新,促进以新能源和新材料为基础的新型工业生物技术的发展。本文针对这些新技术的原理和特点,结合一些典型应用实例,进行分析和总结,希望能为工业微生物的改造与构建提供参考与借鉴。
基因组编辑,寡核苷酸介导的重组,成簇的规律间隔的短回文重复序列,二型内含子,进化工程
近十几年来,生物技术迅猛发展,如高通量的基因组测序技术不断更新换代;基因组信息的指数级增长;高通量分析工具的不断改进,实现了单细胞代谢活性实时定量分析;新型高效的遗传工具和日趋完善的生物信息学分析方法等不断出现,助推了工业微生物育种技术的飞跃式发展[1]。目前,许多工业微生物的育种,不再依赖于过去的模式,如特征表型菌株的筛选、诱变育种和低效的遗传操作技术,而是通过高效的基因组工程改造技术,如对现有菌株的基因组进行人为设计并高效修饰、重组、优化代谢网络,构建新型的工业微生物,高效地制备各种化合物,如甲烷、乙醇、乳酸、丙烯酸、1,3-丙二醇、丁二酸、正丁醇、异丁醇以及1,4-丁二醇等[2]。这些基因组工程技术,改变了以“一次操作、一个抗性基因、一个修饰位点”为特征的繁琐遗传操作模式,以及以“一次改造、一条途径、一个关键基因”为特征的低效途径优化策略,而是实现了在基因组尺度的多重位点和多重途径的组合优化,即基因组编辑 (Genome editing)。
所谓的基因组编辑,是在基因组尺度对细胞进行有效设计与高效改造,如基因组上多个位点同步插入或删除实现多个代谢分支途径的组合优化[3]和外源代谢路径的大片段基因组整合实现全新代谢能力的改造[4]等。这种基因组尺度的高效编辑技术主要包括对基因组上多重位点进行同步改写、高效的插入、替换或删除、大片段的剪切-粘贴以及自主编辑 (即基因组程序性进化) 等。这些技术的应用,哈佛医学院的Church教授进行了形象的比喻,称为对基因组这一密码文本进行的“文本”编辑 (Writing)[5],这体现了人工随意性以及高效性地改造工业微生物基因组时代的到来。
1 基因组多重位点的同步编辑技术
基因组上多重位点的同步编辑对工业微生物代谢性能的改造具有重要意义,比如组合调控多个酶的表达水平,实现分支路径弱化与主代谢路径强化的同步等,克服多重代谢通量优化的细胞代谢网络改造的困难。近几年,不断完善了这种高效的多重基因同步编辑技术,能在基因组范围内对任意的多个位点进行同步修饰[3,6],比如多个基因的RBS序列的修改、基因启动子的同步替换、多个基因的同步失活或删除等等。
1.1 寡核苷酸介导的基因组多重位点编辑技术
基因组的多重位点同步编辑技术首先在大肠杆菌中实现,即基于l-Red重组蛋白Beta的基因组多重位点自动改造技术(Multiplex automated genome engineering,MAGE)[3]。该技术的基本原理是利用人工高效合成的寡核苷酸链 (ssDNA),在噬菌体重组酶介导下,通过同源臂退火到复制叉上单链DNA的目标位点,利用DNA复制过程对基因组进行编辑。多种ssDNA的引入,则实现多重编辑。由于编辑过程可反复循环,不断积累编辑的细胞,最终达到较高的编辑效率,而无需抗生素辅助的筛选过程。这一技术最早源自于中的线性双链DNA高效整合基因组技术[7-8],如图1A所示。将携带有抗性基因和同源臂的双链DNA转化到细胞中时,噬菌体重组酶 (包括l噬菌体来源的Exo/Beta/Gam三种Red蛋白;或Rac噬菌体来源的RecE/RecT) 能够辅助线性DNA整合到基因组同源位点,通过抗性筛选得到突变菌株。不过,该方法一次操作,只能修饰一个基因,且需要抗性筛选,基因组编辑效率较低。后来,对这种噬菌体重组蛋白介导的同源重组的分子机制进行了解析,并革新了这一基因组修饰技术。将含有抗性基因的较长线性双链DNA简化为几十个碱基组成的ssDNA,且多种ssDNA同时转化,可实现多重基因同步编辑。
这种ssDNA介导的基因组编辑技术如图1B所示,其基本步骤是:将人工合成的多种ssDNA混合物转化到感受态细胞中,与胞内过量表达Beta蛋白结合,防止被降解;同时Beta蛋白协助其侵入到基因组复制叉滞后链上的同源位点;ssDNA上的同源臂互补退火后被作为引物合成新生DNA,从而引入突变;主要的突变形式包括点突变和少数碱基的替换、基因插入和基因删除。该方法的典型应用例子是,在中对番茄红素合成途径中的24个关键基因的同步优化[3]。通过多次转化-培养-转化的循环程序,实现了20个基因的RBS序列的随机修改和4个基因删除的组合优化,显著提高产物番茄红素的积累。近几年,由于该方法的高效和通用性,其应用领域被迅速拓展,除了易于遗传转化的革兰氏阴性菌包括大肠杆菌、结核杆菌[9]、鼠伤寒沙门菌、弗氏痢疾杆菌和丁香假单胞菌[10]之外,在转化效率和同源重组频率均偏低的革兰氏阳性菌中也都已被应用,包括枯草芽胞杆菌[11]、罗氏乳杆菌、植物乳杆菌、加氏乳杆菌、乳酸乳球菌[12]以及谷氨酸棒杆菌[13]等。此外,这种ssDNA修饰基因组的方法在真核细胞中也已得到应用,包括酿酒酵母[14]和人体细胞。所以,这种高效的基因组修饰技术必将在未来工业微生物的改造中发挥重要的作用。
1.2 选择标记辅助的多重位点编辑技术
ssDNA介导的基因组编辑技术虽简单高效,但每个位点被改造的碱基数量有限,且筛选过程受细胞转化效率影响较大。为了进一步提高基因组的编辑效率,研究人员新开发了一种有选择标记辅助的MAGE技术 (CoS-MAGE)[6]。该方法在基因组上预先引入一个或多个选择标记的突变基因,通过MAGE同步回复该突变基因作为选择标记,从而筛选到含有其他位点被同步编辑的菌株。这是利用ssDNA在复制叉附近发生多位点同步共编辑的原理,即当细胞处于某个特定复制状态,目标位点被修饰,此时该位点上下游区域发生ssDNA重组事件概率也显著性增强,表现为共修饰现象。利用这种机制,当选择标记基因被ssDNA回复突变时,距离该位点上下游约500 kb的区域内,被同源ssDNA编辑的效率显著提高,一次可实现7–9个位点的同步编辑,且每一个位点的编辑长度也显著增加,比如启动子的替换。所以,CoS-MAGE能够显著性地提高MAGE的效率[15]。此外,由于多个位点的编辑存在一定的概率,在CoS-MAGE的筛选过程中,可同时筛选到不同组合位点被编辑的菌株,从而能够比较不同基因的强弱表达对产物积累的影响[6]。另一种提高MAGE的效率是减弱细胞对ssDNA的降解能力,同时干扰DNA复制过程,增加冈崎片段的长度,从而提高ssDNA修饰效率,该方法也能够进一步提高CoS-MAGE的效率[16]。
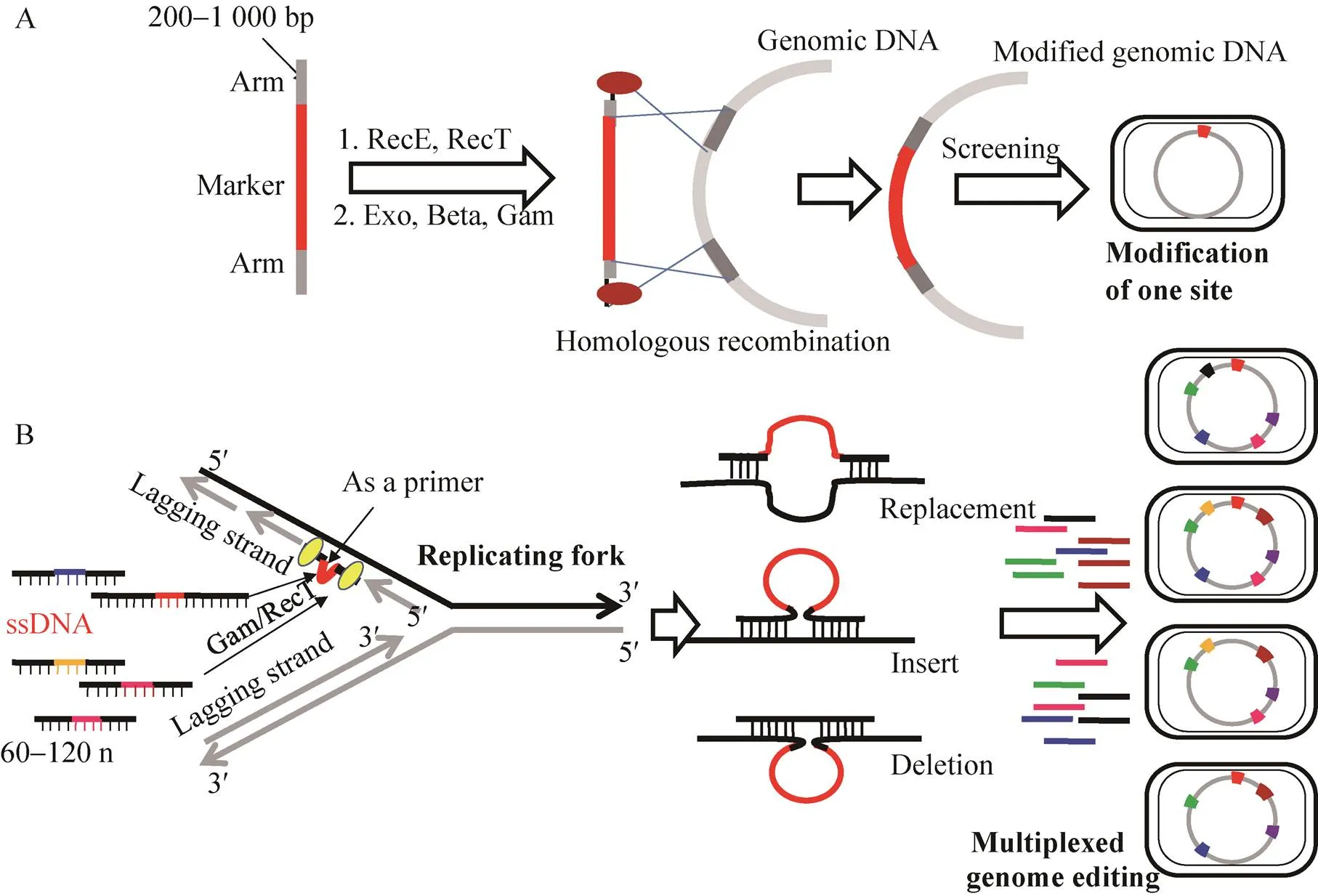
图1 噬菌体重组酶介导的基因组编辑技术
2 双链断裂介导的高效插入、替换和删除
基因组高效编辑技术的另一种策略是,通过自定义人工核酸酶,精确识别并切割基因组特定位点,形成双链断裂,通过胞内的DNA修复系统,实现基因组的编辑。在真核微生物中,双链断裂的DNA修复方式主要包括非同源末端连接 (Non-homologous end-joining,NHEJ) 和同源重组 (Homologous recombination,HR)。其中,NHEJ的方式能够在断裂之处插入或删除少量碱基,其修饰结果是不可控的,适用于特定基因的灭活;而HR的方式则需引入外源同源DNA,可实现目标位点的精确修饰,包括DNA片段的插入、删除和替换,不过其精确性受NHEJ修复方式一定的干扰[17]。由于基因组的双链断裂对细胞毒性较大,所以这种DNA双链断裂介导的基因组编辑效率较高,无需抗生素辅助筛选。不过,这种基因组编辑的操作效率,与人工核酸酶识别DNA序列的改造效率密切相关。近几年,这种技术已得到突飞猛进的发展,革新速度几乎赶超了计算机科技的发展速度,成为了生物科学发展的热点问题。这主要是由于新型核酸酶开发和利用,包括锌指核酸酶 (Zinc-finger nucleases,ZFNs),转录激活因子样效应物核酸酶 (Transcription activator-like effector nucleases,TALENs),归巢核酸内切酶 (Engineered meganucleases) 以及CRISPR-Cas系统 (Clustered regularly interspaced short palindromic repeats- CRISPR-associated system)。
2.1 ZFNs的基因组编辑技术
ZFNs介导的基因组编辑,主要依赖于具备专一性识别DNA序列的锌指结构域 (ZF) 和特异性低的内切核酸酶Ⅰ,如图2A所示。ZFNs识别并切割基因组上的特异性位点,形成双链断裂,极大增加了该位点的重组修复频率,包括非同源末端链接和同源重组,促使切割位点突变产生或将外源引入的DNA片段通过同源重组整合到切割位点。该方法的高效性取决于具有专一性识别DNA位点能力的锌指结构域的构建。该结构域由多个锌指组成,每一个锌指结构域含约30个氨基酸,识别3个碱基对序列,不同锌指组装可识别不同的序列。针对基因组上某一特定基因,需要选择的合适序列,设计特异性识别的ZF,构建和验证ZF与Ⅰ连接产物ZFN的活性,最后用于目标基因的修饰。然而,锌指结构域并不能随机组装,必须借助多种设计方法以及选择优化等[18],比较费时费力;许多被改造的锌指结构域的特异性不高,容易产生脱靶切割。所以,ZFNs在全基因组范围的随机编辑存在一定的局限性。
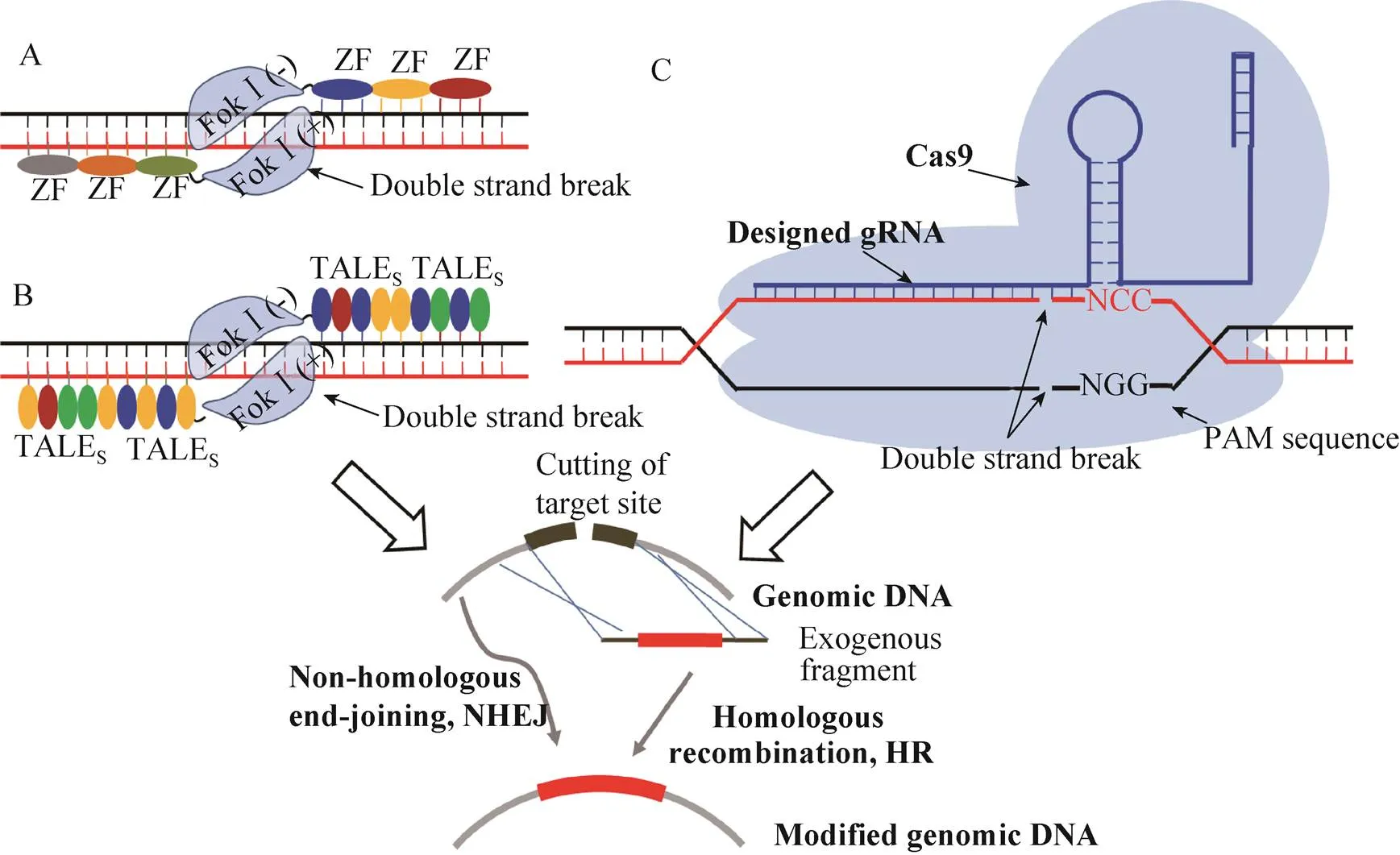
图2 双链断裂介导的基因组编辑技术
2.2 TALENs介导的基因组编辑技术
TALE核酸酶技术于2009年开始被报道,由于其能够克服锌指核酸酶设计上的困难而被广泛关注,在2012年底被期刊评为年度十大突破性技术。该技术的原理是依赖于改造的TALE核酸酶对基因组上特异性位点的识别与切割,从而实现对目的位点的修饰,如图2B所示。天然的TALE蛋白是来源于植物病原菌的一种转录激活效应蛋白,包含有特殊DNA识别结构域和转录激活结构域。大多数TALE蛋白的DNA识别结构域由5–30个重复单元串联而成;一般每个单元由34个氨基酸组成,且高度保守,只有第12和13位的氨基酸高度可变;这些串联的重复单元形成螺旋-转角-螺旋的结构用于识别DNA,且每个重复单元识别一个碱基,由第12和13位的氨基酸决定识别的碱基。由于1个重复单元识别单个碱基,而且不同单元的串联组装对整个结构域的活性影响较小,所以几乎可随机组装,具有很高的适用性。应用高效的组装方法,根据目标DNA序列,自定义组装重复单元,从而极大提升TALE蛋白的DNA识别结构域设计与改造。将TALE蛋白的转录激活结构域替换为非专一性内切核酸酶结构域 (Ⅰ),将其改造为TALE核酸酶,用于基因组编辑。由于Ⅰ的内切核酸酶活性是以二聚体的形式存在,需要构建一对识别比邻DNA的TALE核酸酶,其识别的长度一般是14–20 bp[17]。TALENs编辑基因组的编辑效率往往与这些DNA识别结构域重复单元的组装效率密切相关,目前已有多个研究组构建了一些通用的载体和在线的设计软件[19-20],以及基于磁珠的高通量自动化组装方法[21],使TALENs的效率被不断提升。除了在多细胞真核生物被广泛应用外[17],该技术也已被应用于酵母细胞基因组高效精确的编辑[22]。应用来自水稻黄单胞菌TALE蛋白AvrXa7的4种碱基识别单元,经过PCR结合BⅠ酶切的组装方法进行模块化组装,构建了10个可识别17–23 bp特异性序列的不同TALENs,用于修饰酵母基因组上3个基因的5个不同位点的编辑。结果证实,这些基因位点均被高效修饰,其中通过NHEJ方式的编辑效率在10–4–10–2之间,以基因缺失 (1–70 bp) 造成移码突变为主;若引入外源同源片段,以HR方式进行精确编辑,包括特异性插入失活和基因替换,其效率可提高近100倍,最高的编辑效率达到34%。
2.3 CRISPR-Cas介导的基因组编辑技术
CRISPR-Cas介导的基因组编辑技术是在2013年初才出现,随即被认为是新一代的基因组编辑技术。与TALE核酸酶相比,CRISPR-Cas系统则更加简单方便,易于操作和拓展。CRISPR-Cas系统是许多细菌和古菌都具有的一种防御外源DNA (噬菌体和质粒等) 入侵的原核生物免疫系统。其原理是由CRISPR RNA (crRNA) 识别外源DNA,并被Cas核酸酶切割,所以这是一个由crRNA介导的特异DNA切割系统,如图2C所示。细菌的Ⅱ型CRISPR-Cas系统仅含有单个基因编码的Cas核酸酶Cas9,该蛋白同时具备了crRNA的成熟、特异位点的识别和DNA双链切割的功能,因该系统易于操作而被改造为基因组编辑工具。在天然宿主细胞中,crRNA的成熟需要tracrRNA (trans-acting crRNA) 和RNaseⅢ的辅助,成熟的crRNA与tracrRNA的形成双链RNA后,再与Cas9相互作用形成复合物。此复合物在crRNA上特异序列的指导下,识别特异DNA并在识别位点毗邻的PAM序列之处 (3–9 bp) 进行切割,形成双链断裂。在基因组编辑过程中,这种双链断裂之处同样地可经胞内的两种修复系统进行修复,包括NHEJ和由外源含同源臂DNA辅助的HR,从而实现基因碱基修饰、基因失活、基因删除以及大片段DNA的缺失等[23]。由于crRNA的序列可随意编辑,CRISPR-Cas9系统的基因组编辑技术被认为适用于基因组上任意位点的精确编辑。
在实际应用中,crRNA与tracrRNA形成的双链可以采用人工设计的gRNA (Guide RNA) 取代 (图2C),gRNA自主折叠形成Cas9可识别的双链RNA,简化了CRISPR-Cas9系统。该系统已被应用于细菌[24]、酵母[25]、植物、线虫、石斑鱼、老鼠以及人的细胞等。其中在酿酒酵母中,采用人工设计的gRNA,在Cas9的辅助下,基因组的定点突变和同源基因替换的效率接近100%,实现了简单、高效、精确的基因组编辑[25]。除了对基因组DNA定点编辑之外,通过对Cas9的改造,消除其核酸内切酶的活性 (dCas9),改造的CRISPR-dCas9系统可应用于单个或多重基因的表达调控,包括细菌和真核生物[26-27]。其原理是:1) 在细胞内表达gRNA后,使其与dCas9形成复合物,识别目标基因但不发生双链切割,可阻止该基因的转录延伸、核糖体结合或是转录因子结合,使基因表达水平显著下降,一些基因的转录水平甚至下降至原来的10–4水平,几乎沉默该基因的表达[27];2) 在dCas9蛋白上融合转录激活结构域或抑制结构域,由表达的gRNA精确引导,使其结合到目标基因的调控区,实现稳定且精确地调控基因表达[26]。通过引入多重gRNA,可实现多重基因的表达调控,具有简单、高效和精确的特点[27]。
3 基因组大片段的插入、删除和剪切- 粘贴
在基因组范围进行自定义编辑,大片段基因的操作是必不可少的,比如一整条次级代谢途径的整合、一些非必需基因的大片段删除等。尽管自定义人工合成基因组是最简单直接的,但是其成本太高,难以被推广应用。在现有基因组的基础上,采用低成本的大片段合成DNA,并进行自定义改造则更容易操作,可提高基因组大片段编辑的效率。这种大片段DNA插入或删除技术主要是由位点专一的重组酶和它的特异性识别位点组成,包括最早被识别的l噬菌体溶源性基因组整合系统:整合酶和专一性位点 (自身基因组上的位点和宿主基因组上的位点);P1噬菌体的Cre/重组系统;酵母的Flp/FRT重组系统等。
3.1 位点专一性重组酶的基因组编辑
应用这些重组酶进行基因组编辑,其原理是在基因组上和目的DNA上均插入特异性识别序列,在相应的重组酶作用下进行重组,实现大片段DNA的插入、删除或替换,如图3所示。比如基因替换操作:在基因组上目的片段两端分别插入两个重组酶识别位点;同样地,外源片段含有相应重组酶识别位点;通过重组酶的过量表达,即可实现两个特异识别位点之间基因组DNA的替换操作。而将3个不同的识别位点组合使用,则可以在基因组上反复插入大片段DNA[28]。这种重组酶介导的大片段编辑效率高、专一性好,而且宿主通用性强。但是仍存在一个瓶颈问题,即重组酶的特异性识别序列难以自定义改造,所以必须在基因组中预先引入重组酶的识别位点,这往往需要借助其他方法。最新研究的一种策略是将位点专一重组和二型内含子归巢相结合[29],这种方法能有效地解决这个瓶颈问题,显著提高基因组上大片段改造的效率。
3.2 二型内含子归巢介导的基因组编辑技术
二型内含子归巢是近几年发展的一种高效基因组插入技术,具有较好的通用性,已被广泛地应用于各种宿主细胞,包括一些遗传操作比较困难的细菌,如梭菌[30]。该技术也已被Sigma公司商业化,用于自定义高效编辑不同细菌的基因组。被研究最多的是来源于乳酸乳球菌的Ll.LtrB内含子,此外最近还开发一个新的来源于的EcI5内含子[31]。这些内含子的归巢原理如图3所示:当内含子经过外源质粒的引入,并转录成为RNA时,折叠成为具有特殊三级结构的功能RNA;在自身编码的一个多功能辅助蛋白的帮助下,内含子上两个特异性识别位点 (EBS1和EBS2) 被相互靠近,用于互补识别基因组上的相互靠近的两个特异位点 (E1和E2);三级结构的内含子RNA核酶切割并连接DNA链,由辅助蛋白将其逆转录为编码的DNA;在胞内DNA修复系统帮助下,完成内含子DNA的插入归巢。二型内含子的插入特异性由EBS1和EBS2决定,而这两个位点的序列可根据一定的算法进行修改,从而特异性识别基因组上不同的位点,几乎可实现基因组的随机编辑[32]。在一些微生物中,这种内含子介导的基因组插入效率可高达80%,且无需抗性筛选标记[33]。
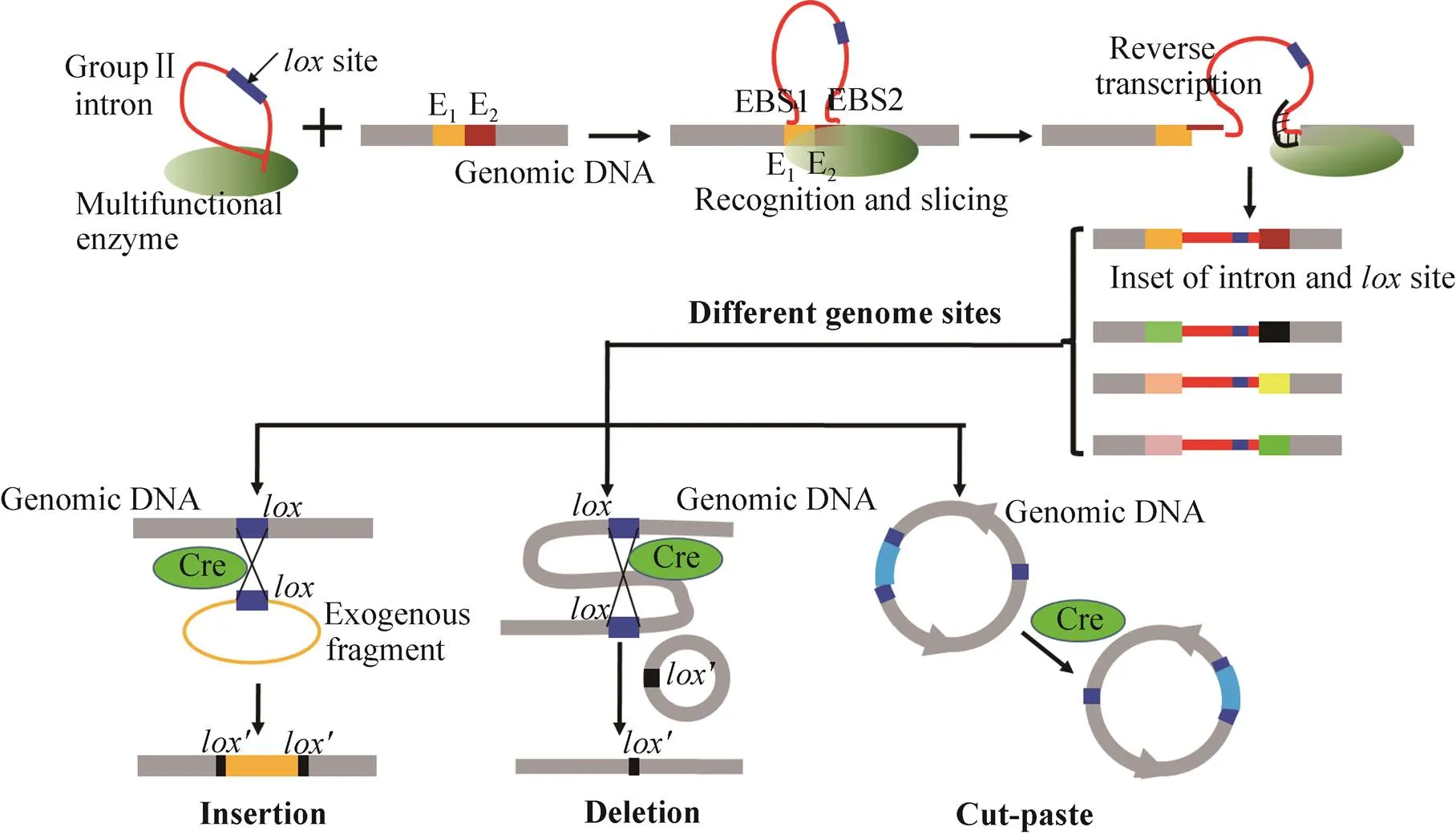
图3 基因组上大片段DNA的高效编辑技术[29]
3.3 基因组大片段DNA高效编辑技术
将二型内含子介导的高效定点插入技术与位点专一性重组介导的大片段DNA编辑技术相结合,被称为GETR (Genome editing via targetrons and recombinases) 技术[29]。GETR能够在基因组上多重位点进行大片段DNA的插入、删除、翻转以及剪切-粘贴等操作,包括在中插入12 kb的聚酮合成操纵子或剪切-粘贴120 kb的基因组、在金黄色葡萄球菌中多个区域同时删除120 kb的基因组、在枯草芽胞杆菌中翻转 1.2 Mb的基因组等。而且这种大片段基因组修饰过程无需选择性标记,其效率接近100%。其基本原理如图3所示:将二型内含子Ll.LtrB或EcI5进行改造,使改造的内含子包含有重组酶Cre的特异性识别位点位点,并恢复内含子的归巢效率;通过改造的内含子,能够将不同位点高效地插入到基因组的多个目标位置;通过Cre的重组作用,实现基因组上大片段的高效编辑。
4 基因组自主编辑的新策略
局限于对细胞系统的有限认识,并非所有的细胞特性都能通过理性的技术进行改造,比如细胞的耐受性[34]。而生物系统是一个自主装系统,经过长期的进化和选择,形成了一个鲁棒的生物细胞,使细胞各个元件能够协同作用,适应胁迫环境并获得竞争优势,所以微生物基因组系统内存在的进化能力,可进行自主编辑,提升自身系统[35-36]。应用基因组进化,近几年也开发了一些新的基因组进化技术,实现基因组自主编辑,如图4所示,比如结合原生质体融合的基因组重排技术 (Genome shuffling)[37]、改造基因组范围转录调控程序的全局转录因子工程 (Global transcription machinery engineering, gTME)[38]、基因组复制机器工程辅助的连续进化(Genome replication engineering assisted continuous evolution,GREACE)[39]以及环境条件诱导的适应性进化策略 (Stress-induced-mutagenesis (SIM) based adaptive evolution)[40]等。
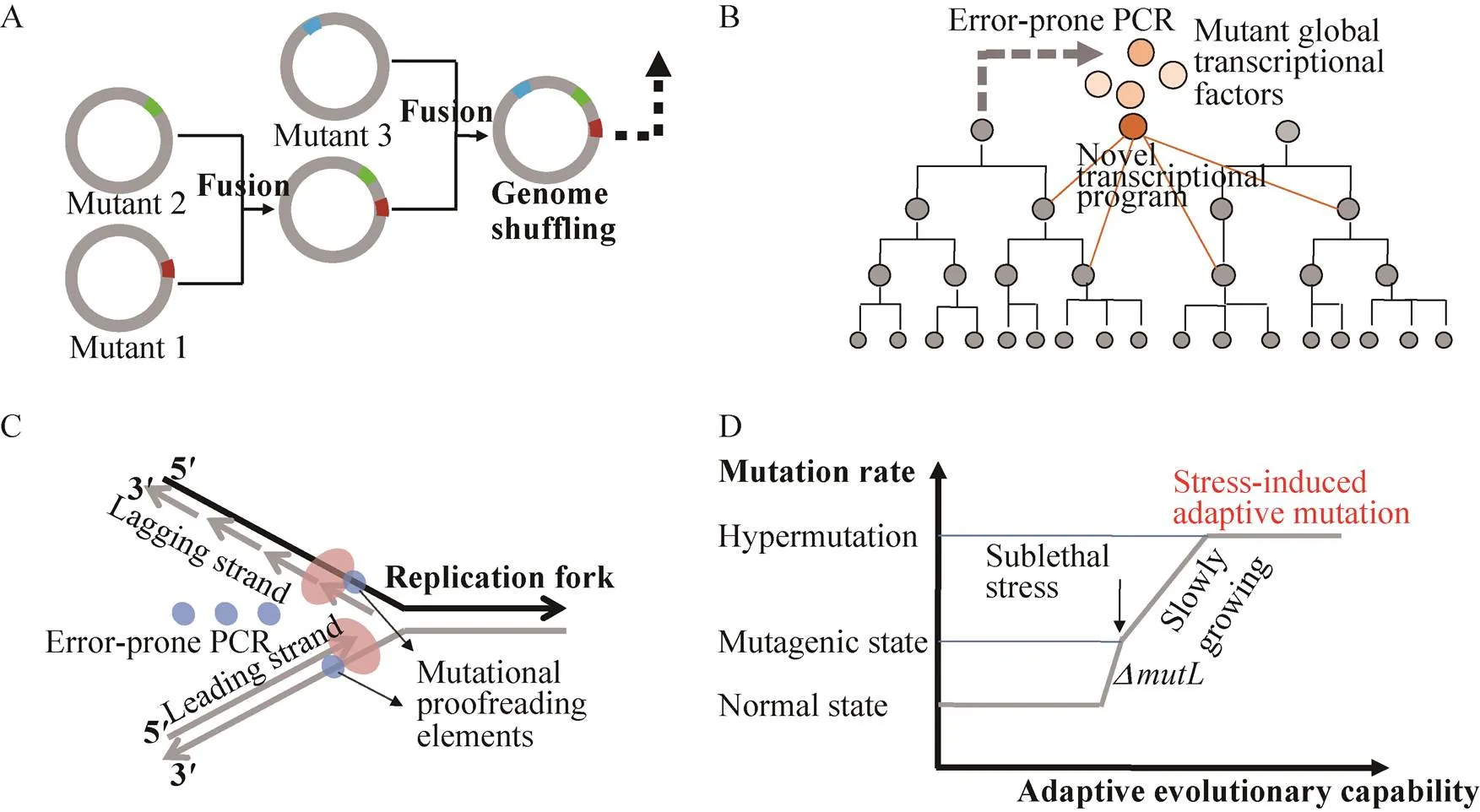
图4 新发展的微生物基因组进化技术
这些基因组自主编辑技术具有不同的特点,并能够有效改造微生物细胞的复杂表型:
1) 基因组重排技术 (图4A),是将传统的诱变育种技术 (获得丰富多样性表型的出发突变株文库)、原生质体融合技术 (不同突变株间基因组发生重排) 与高通量筛选技术相结合,使诱变过程中产生的丰富的基因组 (多种突变表型),由原生质体融合而发生大片段重排。随后,经过选择,使多种正向突变在融合菌株中积累,加速获得目的性状的突变菌株,消除传统诱变步步积累的繁琐、低效以及积累大量有害突变的缺点。该技术的首次应用是提高乳酸生产菌的低pH耐受性[37]。首先是通过两种方法获得的表型多样性的起始亲本突变菌株,包括恒化培养 (驯化) 和NTG诱变;再者,经过5轮递归原生质体融合,最终筛选到了5株低pH耐受性显著提高的乳酸生产菌。通过两个营养缺陷型的标记证实了耐受性提高的菌株发生了基因组重排。
2) 全局转录因子工程 (图4B),是结合定向进化的方法,突变全局转录因子,使细胞的转录程序在基因组水平上发生随机变化。也就是定向进化 (如易错PCR方法) 核心转录因子,随机改变细胞转录机器的转录偏好性和转录能力,形成差异丰富多样的转录程序,筛选最优的转录程序,达到全细胞范围的转录程序优化的目的。如酿酒酵母中[38],通过对RNA聚合酶中的TATA框结合蛋白 (SPT) 或TATA框结合辅助蛋白 (TAF) 进行多轮的定向进化和筛选后,优化了酿酒酵母抵抗高浓度乙醇的全细胞基因转录程序。如突变体SPT15,显著提高了酵母在高浓度葡萄糖下的生长速率和对高浓度乙醇的耐受能力,使菌株的乙醇生产速率提高70%。基因功能分析表明,突变的全局转录因子SPT15,使细胞中上百个基因的表达发生变化,正是这些基因的组合效果赋予了细胞新的表型,而非单个或多个组合的基因变化。
3) 基因组复制机器工程辅助的连续进化 (图4C),是最新开发的一种条件性改变基因组复制机器的保真性,增加基因组突变率,从而能够更有效地突变并筛选目的表型的菌株。即通过多轮的易错PCR,随机改造DNA聚合酶中的校正亚基,并将其转化到宿主细胞中,改变DNA聚合酶复制基因组时的保真性,通过筛选和富集,获得突变菌株。例如在改造的进化能力时[39],对DNA聚合酶的亚基进行易错PCR的定向进化,并转化细胞,使其在胁迫环境中进行多轮的连续进化并筛选,最终获得耐受性显著提高的突变菌株,包括抗生素的抗性、高浓度乙酸或丁醇的耐受性。该方法操作简单方便、通用性强,只需引入一个DNA聚合酶中的校正功能的突变亚基;而且易于扩展,GREACE可用于提高工业菌株对高浓度底物和高浓度产物耐受性等复杂表型。
4) 结合环境条件诱导作用的适应性进化策略 (图4D)。其显著特点是采用生长缓慢或者不生长的细胞,应用细胞自身受环境胁迫而大幅提升的内在进化动力[41],加速细胞的适应性进化;同时实现单细胞突变与筛选的同步,不但减少筛选的过程,也简化操作时间,能够快速地获得突变菌株[40]。应用该方法,经过12轮的突变筛选,能够将的丁醇最小抑制浓度从最初的9.5 g/L提高到13 g/L;而通过5轮的突变筛选,的高渗透压耐受性和温度耐受性也被显著提升。所以,应用基因组内在的诱导性进化动力,结合环境胁迫因素,能够提升基因组自主编辑的效率,用于改造一些复杂的生理表型。
5 结论
结合高通量的DNA合成技术和高效的遗传操作新工具,基因组编辑技术不断推陈出新,能更有效地解决工业微生物改造中的许多瓶颈问题,包括基因组上多重位点的同步组合优化、无需抗生素辅助筛选的高效基因组修饰、大片段基因组的改造和复杂表型的改造等。新的基因组编辑技术的应用,极大提升了工业菌种的改造速度,现已成为应用研究领域的一大热点。如Genomatica、Codexis、LS9、Genencor等为代表的一批公司已在这方面积累核心技术。Genomatica利用自身发展起来的基因组工程技术和基因组网络改造策略,对1,4-丁二醇、丙烯酸、异丙醇等化工产品的生物制造路线申请了超过70份概念型专利。由此可见,工业微生物基因组编辑技术将在未来几年得到广泛应用。例如,对传统发酵工业中的重要微生物进行基因组改造,构建全新的生产菌株替代传统菌株,如氨基酸、有机酸、抗生素、维生素、色素等,提升或淘汰现有生产方式;构建新型的合成代谢网络和工业生产菌株以及新型的微生物生态群落,实现新型能源和新材料的生物制造等。但这些技术的快速发展也面临一些问题。如一些高效的基因组编辑技术只能在一些容易遗传操作的模式菌中应用,难以应用到其他微生物;工业菌株的改造靶点,需要由较为成熟的计算机工具辅助,这些方面仍需要不断地完善。
[1] Keasling JD. Manufacturing molecules through metabolic engineering. Science, 2010, 330(6009): 1355–1358.
[2] Yu B, Zhang XL, Li Y, et al. Cell factories for biorefinery. China Basic Sci, 2009, 5: 14–19 (in Chinese).于波, 张学礼, 李寅, 等. 生物炼制细胞工厂的科学基础. 中国基础科学, 2009, 5: 14–19.
[3] Wang HH, Isaacs FJ, Carr PA, et al. Programming cells by multiplex genome engineering and accelerated evolution. Nature, 2009, 460(7257): 894–898.
[4] Kuhlman TE, Cox EC. Site-specific chromosomal integration of large synthetic constructs. Nucleic Acids Res, 2010, 38(6): e92.
[5] Church GM. Reading and writing omes. Mol Syst Biol, 2013, 9: 642.
[6] Wang HH, Kim H, Cong L, et al. Genome-scale promoter engineering by coselection MAGE. Nat Methods, 2012, 9(6): 591–593.
[7] Zhang YM, Buchholz F, Muyrers JPP, et al. A new logic for DNA engineering using recombination in. Nat Genet, 1998, 20(2): 123–128.
[8] Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes inK-12 using PCR products. Proc Natl Acad Sci USA, 2000, 97(12): 6640–6645.
[9] van Kessel JC, Hatfull GF. Recombineering in. Nat Methods, 2007, 4(2): 147–152.
[10] Swingle B, Markel E, Costantino N, et al. Oligonucleotide recombination in Gram-negative bacteria. Mol Microbiol, 2010, 75(1): 138–148.
[11] Wang Y, Weng J, Waseem R, et al.genome editing using ssDNA with short homology regions. Nucleic Acids Res, 2012, 40(12): e91.
[12] van Pijkeren JP, Britton RA. High efficiency recombineering in lactic acid bacteria. Nucleic Acids Res, 2012, 40(10): e76.
[13] Binder S, Siedler S, Marienhagen J, et al. Recombineering incombined with optical nanosensors: a general strategy for fast producer strain generation. Nucleic Acids Res, 2013, 41(12): 6360–6369.
[14] DiCarlo JE, Conley AJ, Penttila M, et al. Yeast oligo-mediated genome engineering (YOGE). ACS Synth Biol, 2013, 2(12): 741–749.
[15] Carr PA, Wang HH, Sterling B, et al. Enhanced multiplex genome engineering through co-operative oligonucleotide co-selection. Nucleic Acids Res, 2012, 40(17): e132.
[16] Lajoie MJ, Gregg CJ, Mosberg JA, et al. Manipulating replisome dynamics to enhance lambda red-mediated multiplex genome engineering. Nucleic Acids Res, 2012, 40(22): e170.
[17] Wei C, Liu J, Yu Z, et al. TALEN or Cas9-rapid, efficient and specific choices for genome modifications. J Genet Genomics, 2013, 40(6): 281–289.
[18] Sander JD, Dahlborg EJ, Goodwin MJ, et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat Methods, 2011, 8(1): 67-69.
[19] Zhang F, Cong L, Lodato S, et al. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat Biotechnol, 2011, 29(2): 149–153.
[20] Li L, Piatek MJ, Atef A, et al. Rapid and highly efficient construction of TALE-based transcriptional regulators and nucleases for genome modification. Plant Mol Biol, 2012, 78(4/5): 407–416.
[21] Reyon D, Tsai SQ, Khayter C, et al. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol, 2012, 30(5): 460–465.
[22] Li T, Huang S, Zhao XF, et al. Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res, 2011, 39(14): 6315–6325.
[23] Segal DJ. Bacteria herald a new era of gene editing. Elife, 2013, 2: e00563.
[24] Jiang WY, Bikard D, Cox D, et al. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol, 2013, 31(3): 233–239.
[25] DiCarlo JE, Norville JE, Mali P, et al. Genome engineering inusing CRISPR-Cas systems. Nucleic Acids Res, 2013, 41(7): 4336–4343.
[26] Gilbert LA, Larson MH, Morsut L, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell, 2013, 154(2): 442–451.
[27] Qi LS, Larson MH, Gilbert LA, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell, 2013, 152(5): 1173–1183.
[28] Turan S, Galla M, Ernst E, et al. Recombinase-mediated cassette exchange (RMCE): traditional concepts and current challenges. J Mol Biol, 2011, 407(2): 193–221.
[29] Enyeart PJ, Chirieleison SM, Dao MN, et al. Generalized bacterial genome editing using mobile group II introns and Cre-lox. Mol Syst Biol, 2013, 9: 685.
[30] Heap JT, Pennington OJ, Cartman ST, et al. The ClosTron: a universal gene knock-out system for the genus. J Microbiol Meth, 2007, 70(3): 452–464.
[31] Zhuang FL, Karberg M, Perutka J, et al. EcI5, a group IIB intron with high retrohoming frequency: DNA target site recognition and use in gene targeting. Rna, 2009, 15(3): 432–449.
[32] Perutka J, Wang WJ, Goerlitz D, et al. Use of computer-designed group II introns to disruptDExH/D-box protein and DNA helicase genes. J Mol Biol, 2004, 336(2): 421–439.
[33] Yao J, Zhong J, Lambowitz AM. Gene targeting using randomly inserted group II introns (targetrons) recovered from angene disruption library. Nucleic Acids Res, 2005, 33(10): 3351–3362.
[34] Zhang Y, Zhu Y, Li Y. The importance of engineering physiological functionality into microbes. Trends Biotechnol, 2009, 27(12): 664–672.
[35] Zhu L, Zhu Y, Zhang Y, et al. Engineering the robustness of industrial microbes through synthetic biology. Trends Microbiol, 2012, 20(2): 94–101.
[36] Kitano H. Towards a theory of biological robustness. Mol Syst Biol, 2007, 3: 137.
[37] Zhang YX, Perry K, Vinci VA, et al. Genome shuffling leads to rapid phenotypic improvement in bacteria. Nature, 2002, 415(6872): 644–646.
[38] Alper H, Moxley J, Nevoigt E, et al. Engineering yeast transcription machinery for improved ethanol tolerance and production. Science, 2006, 314(5805): 1565–1568.
[39] Luan GD, Cai Z, Li Y, et al. Genome replication engineering assisted continuous evolution (GREACE) to improve microbial tolerance for biofuels production. Biotechnol Biofuels, 2013, 6(1): 137.
[40] Zhu L, Cai Z, Zhang Y, et al. Engineering stress tolerance ofby stress-induced- mutagenesis (SIM) based adaptive evolution. Biotechnol J, 2014, 9(1): 120–127.
[41] Zhu L, Li Q. Stress-induced cellular adaptive mutagenesis. Hereditas (Beijing), 2014, 36(4): 327–335 (in Chinese).朱林江, 李崎. 环境胁迫诱导的细胞适应性突变. 遗传, 2014, 36(4): 327–335.
(本文责编 郝丽芳)
Genome editing of industrial microorganism
Linjiang Zhu1,2, and Qi Li1,2
1 The Key Laboratory of Industrial Biotechnology, Ministry of Education, School of Biotechnology, Jiangnan University, Wuxi 214122, Jiangsu, China2 Synergetic Innovation Center of Food Safety and Nutrition, Jiangnan University, Wuxi 214122, Jiangsu, China
Genome editing is defined as highly-effective and precise modification of cellular genome in a large scale. In recent years, such genome-editing methods have been rapidly developed in the field of industrial strain improvement. The quickly-updating methods thoroughly change the old mode of inefficient genetic modification, which is “one modification, one selection marker, and one target site”. Highly-effective modification mode in genome editing have been developed including simultaneous modification of multiplex genes, highly-effective insertion, replacement, and deletion of target genes in the genome scale, cut-paste of a large DNA fragment. These new tools for microbial genome editing will certainly be applied widely, and increase the efficiency of industrial strain improvement, and promote the revolution of traditional fermentation industry and rapid development of novel industrial biotechnology like production of biofuel and biomaterial. The technological principle of these genome-editing methods and their applications were summarized in this review, which can benefit engineering and construction of industrial microorganism.
genome editing, ssDNA-mediated recombination, clustered regularly interspaced short palindromic repeats, group-II intron, evolutionary engineering
June 18, 2014;Accepted:September 15, 2014
Qi Li. Tel: +86-510-85918176; E-mail: liqi@jiangnan.edu.cn
Supported by:the Fundamental Research Funds for the Central Universities (Nos. JUSRP51306A, JUDCF13008, JUSRP51402A), Jiangnan University Basic Research Foundation for Young Scholars (No. 1012050205134030), the Priority Academic Program Development of Jiangsu Higher Education Institutions, the 111 Project (No. 111-2-06), the Jiangsu Province “Collaborative Innovation Center for Advanced Industrial Fermentation” Industry Development Program.
中央高校基本科研业务费资金 (No. JUSRP51306A, JUSRP51402A, JUDCF13008),江南大学基本科研-青年基金项目(No. 1012050205134030),江苏高校优势学科建设工程资助项目,111引智计划 (No. 111-2-06),江苏省现代发酵工业协同创新中心资助。
2014-10-14
http://www.cnki.net/kcms/doi/10.13345/j.cjb.140340.html
猜你喜欢
内蒙古师范大学学报(自然科学汉文版)(2021年3期)2021-06-01
无机化学学报(2020年7期)2020-07-20
中国预防兽医学报(2020年2期)2020-06-01
三农资讯半月报(2020年8期)2020-05-13
生物工程学报(2019年6期)2019-07-10
广州大学学报(自然科学版)(2019年1期)2019-05-07
生物学通报(2019年1期)2019-02-15
生物学通报(2018年12期)2018-10-10
中国病理生理杂志(2017年2期)2017-01-17
天津科技大学学报(2016年1期)2016-02-28
