
Baeyer-Villiger单加氧酶非保守Hinge影响酶的催化活性和立体选择性
2015-07-19 13:08梁秋玲吴胜
生物工程学报 2015年3期
梁秋玲,吴胜
Baeyer-Villiger单加氧酶非保守Hinge影响酶的催化活性和立体选择性
梁秋玲1,2,吴胜1
1 中国科学院微生物研究所微生物资源前期开发国家重点实验室,北京 100101 2 中国科学院大学,北京 100049

梁秋玲, 吴胜. Baeyer-Villiger单加氧酶非保守Hinge影响酶的催化活性和立体选择性. 生物工程学报, 2015, 31(3): 361–374.Liang QL, Wu S. Nonconserved hinge in Baeyer-Villiger monooxygenase affects catalytic activity and stereoselectivity. Chin J Biotech, 2015, 31(3): 361–374.
Baeyer-Villiger单加氧酶是一种重要的生物催化剂,可用于合成一系列有价值的酯和内酯化合物。通过序列比对和晶体结构分析推测连接NADPH结构域和FAD结构域的一段非保守Hinge可能在酶对底物识别和催化氧化过程中扮演着重要角色。在以环己酮单加氧酶为模型的研究中发现,对该Hinge结构进行同源序列替换得到的突变体几乎完全丧失了催化活性,证明了其整体水平的重要性。丙氨酸扫描突变揭示其中一些位点对酶的功能有显著影响:K153位点的改变使酶的活性下降,立体选择性却更优化;L143位点的改变对酶的活性影响较小,却降低了立体选择性;L144位点的改变则同时大幅度削弱酶的活性和立体选择性。将同样的方法运用在苯丙酮单加氧酶中,我们得到了相似的结论,证明这些位点的重要功能在Baeyer-Villiger单加氧酶家族中有一定的普遍性。这一研究增进了对Baeyer-Villiger单加氧酶的结构与功能关系的认识,有助于底物结合口袋的精确描述和Baeyer-Villiger单加氧酶催化图景的进一步细化,对未来相关的理性设计和定向改造研究提供了借鉴。
Baeyer-Villiger单加氧酶,环己酮单加氧酶,同源替换,丙氨酸扫描突变
Baeyer-Villiger氧化反应是一类重要的有机化学反应,它能实现功能基的转化,活化C-C键进行环扩张,进而合成一系列有价值的手性酯和内酯化合物[1]。这一反应在1899年就被首次报道[2],人们也一直致力于研究使用各种有效的催化剂来实现这一反应[3]。但是由于化学催化剂本身固有的弱点,催化效率低、选择性低、污染以及反应过程复杂副产物多等,研究者们开始向生物酶催化领域寻找更优越的催化剂。
早在1976年,第一个Baeyer-Villiger单加氧酶CHMO就已经被发现和分离[4],此后又有许多各种不同类型的BVMO被发现。本世纪初相继有类固醇单加氧酶 (Steroid monooxygenase, STMO)[5]、对羟基苯乙酮(4-Hydroxyacetophenone monooxygenase)[6]、环戊酮单加氧酶(Cyclopentanone monooxygenase, CPMO)[7-8]等新型BVMO的报道。2002年,Ⅰ型BVMO的特异性序列被首次提出[9],以此为基础,人们通过基因组挖掘 (Genome mining) 的方法,在细菌和真菌基因组中都发现大量Ⅰ型BVMO特异性序列的分布,在动物和植物细胞中则不存在Ⅰ型BVMO[10]。近年来,关于BVMO多样性的报道越来越多[11-12],有许多BVMO都在微生物代谢途径中起着重要作用[13-14]。
这些酶的发现无疑给涉及Baeyer-Villiger单加氧反应的工业生产和药物生产带来了革新的希望[15-16]。事实上,许多研究证明,BVMO还在许多其他领域表现出很好的运用价值,例如,潜在的药物靶点设计[17]、潜在的基团保护方式等[18]。因为其广泛的作用,近年来,人们对各类新型BVMO从晶体结构、催化机制、底物谱和立体选择性等方面进行了大量的研究,也取得了一系列重要突破。其中具有划时代意义的是2004年PAMO晶体结构的首次提出[19]。PAMO晶体结构由两个结构域组成:FAD和NADPH结构域,其催化活性位点位于晶体表面的一个凹缝处。2009年,又有第2个BVMO的晶体结构被解析出[20],揭示了CHMO两种不同状态的晶体结构。“开”、“闭”两种结构的展示进一步细化了BVMO的催化图景,说明BVMO催化反应是一个动态的过程,辅基会在其催化位点发生位移。同年还发现了另一种不依赖于FAD和NADPH的BVMO——MtmOIV的晶体结构,这一类BVMO被称为Ⅱ型BVMO[21-22]。2012年,Yachnin等[23]首次报道了CHMO与底物小分子环己酮相结合的晶体结构,向我们清晰地展示了BVMO催化过程中与底物结合形成的重要中间体的形态。这些晶体的解析有助于深入理解这一类酶的底物识别及催化机制,并为酶的分子改造提供重要的结构信息。近些年来,结合结构信息和实验室定向进化的手段建立突变酶库,并从中筛选出最适用于生产应用的突变株已经成为BVMO的研究热点。其中研究背景最悠久的CHMO和热稳定性最好的PAMO成为研究工作者们青睐的对象。
蛋白晶体的解析、结构信息的阐释以及众多分子定向进化的成功例子都让我们对BVMO的底物识别、催化机制等有了更深的认识。但是催化过程中的位移细节乃至催化的精确机制至今未知,包括底物和氧气分子的激活、中间体如何形成、中间体如何稳定等。特别是这些过程如何精细调控了各种BVMO独特的底物特异性和立体选择性还有待进一步深入研究。为了探索这些问题的答案,本研究首先对BVMO的基因序列和晶体结构进行深入分析,在底物结合口袋附近发现了一段连接NADPH结构域和FAD结构域在BVMO催化过程中形态难以固定的区域,即HingeⅠ结构。迄今为止,对Hinge的认识多数还停留在结构数据支撑上,并没有系统地对酶催化功能影响的研究,少数研究也只是浅尝辄止[24-25]。本课题以CHMO为模型,旨在探索非保守Hinge对酶催化行为的影响,通过改进的Quick-change和overlap PCR方法获得一系列CHMO突变酶,包括两个HingeⅠ同源序列替换突变体CHMOPAMO、CHMOSTMO以及HingeⅠ序列的丙氨酸扫描突变体。通过对突变体进行酶的活性检测和立体选择性的研究,得到了一些影响CHMO活性和选择性的重要位点,证明了HingeⅠ这一结构在CHMO中乃至整个BVMO家族的催化过程中都起着重要作用。这一研究有助于BVMO催化图景的进一步细化,给我们未来对BVMO理性进化设计和定向改造研究提供借鉴。
1 材料与方法
1.1 材料
1.1.1 工具酶与试剂
PCR所用的Ex酶以及T载体质粒扩增系统、KODDNA聚合酶、限制性内切酶、DNA连接酶分别购自TaKaRa公司(TaKaRa Bio Group)、日本Toyobo公司、NEB (New England Biolabs) 公司以及MBI (MBI Fermentas) 公司;质粒提取试剂盒、DNA凝胶电泳回收试剂盒购自天根生化科技 (北京) 有限公司;阿拉伯糖购自北京赛百盛基因技术有限公司;蛋白胨、酵母膏、氯化钠均为国产分析纯试剂。
1.1.2 菌株与质粒
大肠杆菌Top10、BL21(DE3) Gold以及质粒pBAD、pET22b均为本实验室保存。
1.1.3 培养基
LB培养基:蛋白胨10 g,酵母提取物5 g,氯化钠10 g,蒸馏水定容1 L,pH调为7.0,121 ℃高压蒸汽灭菌20 min,固体培养基时加1.5%琼脂。TB培养基:蛋白胨12 g,酵母提取物24 g,甘油4 mL,各组分溶解后高压灭菌,冷却到60 ℃,再加入100 mL灭菌的170 mmol/L KH2PO4和 0.72 mol/L K2HPO4的溶液 (2.31 g的KH2PO4和12.54 g K2HPO4溶在足量的水中,使终体积为100 mL),121 ℃高压蒸汽灭菌20 min。
1.2 方法
1.2.1 突变体构建
PAMO/CHMO/STMO的基因序列分别来自NCBI数据库 (http://www.ncbi.nlm.nih.gov/) 的噬热裂孢菌(AAZ55526),不动杆菌sp. NCIMB9871 (BAA86293) 和玫瑰色红球菌(BAA24454)。
同源序列替换突变体构建:将CHMO的HingeⅠ序列分别替换成PAMO和STMO的HingeⅠ序列,得到突变体CHMOPAMOand CHMOSTMO。具体替换过程见补充材料2 (SM2)。CHMOPAMO和CHMOSTMO被连接到pET22b中,最后将重组质粒转入BL21(DE3) Gold中进行蛋白表达。
丙氨酸扫描突变体构建:使用Quick-change PCR[24]方法构建以下突变体:1) CHMO: Leu143Ala/Leu144Ala/Ser145Ala/Pro147Ala/Asn148Ala/Leu149Ala/Pro150Ala/Leu151Ala/Ile152Ala/Lys153Ala; 2) PAMO: Gln152Ala/Leu153Ala/Ser1 54Ala/Val155Ala/Pro156Ala/Gln157Ala/Leu158Ala/Pro159Ala/Asn160Ala/Phe161Ala/Pro162Ala。
PCR反应体系为25 μL终体积:10×KOD缓冲液 (2.5 μL),MgCl2(1 μL, 0.025 mol/L),dNTPs (5 μL,每种核苷酸浓度为0.002 mol/L),引物 (0.5 μL,正反引物浓度均为2.5 μmol/L),模板质粒 (1 μL,10 ng/μL) 和1单位KOD DNA聚合酶。PAMO的PCR反应循环条件为:94 ℃ 10 min;94 ℃变性40 s,53 ℃退火40 s以及72 ℃延伸50 s,循环35次;94 ℃变性1 min,60 ℃退火1 min以及72 ℃延伸7 min,循环35次;最后在72 ℃下延伸30 min。CHMO的PCR条件中,第2次循环的退火温度为56 ℃,延伸时间为8 min,其他参数与PAMO相同。
PCR之后取10 μL PCR产物,加入1.2 μL NEB缓冲液,1 μL DpnⅠ,37 ℃处理3 h。最后转入Top10 或者BL21(DE3) Gold宿主菌。
1.2.2 突变体基因表达
重组质粒pET22b-CHMO转入BL21(DE3) Gold,细胞在含有100 μg/mL氨苄青霉素的LB培养基中37 ℃过夜培养。每5 mL过夜培养的培养基再转入200 mL含有100 μg/mL氨苄青霉素的TB培养基,37 ℃培养至600到达0.6,此时加入终浓度为25 μmol/L IPTG (Isopropyl-β-D-thiogalactopyranoside) 诱导,并将培养温度调至20 ℃,转速调至150 r/min,培养12 h。最后,细胞以0.9% NaCl洗涤收集。重组质粒pBAD-PAMO转入Top10中,5 mL过夜培养物转入200 mL含有0.1%阿拉伯糖诱导剂和100 μg/mL氨苄青霉素的TB培养基中,37 ℃、220 r/min培养20 h。最后细胞同样以0.9% NaCl洗涤收集。这两种重组细胞都将用于全细胞生物催化和酶活检测。
1.2.3 全细胞催化
通过测定600的方法将表达不同突变蛋白的细胞调成相同浓度,然后将细胞悬浮于10 mL缓冲液 (20 mmol/L Tris-HCl,pH 8.0) 中。取其中500 μL,放入终浓度为1 mmol/L底物进行反应,PAMO对绝大部分底物反应温度设置为35 ℃,芳香环酮类底物反应温度设置为37 ℃。CHMO反应温度均为32 ℃。所有反应均在 13 000 r/min的振荡器中完成,反应时间控制在12 h (CHMO) 或者2 h (PAMO) (PAMO与芳香环酮类底物的反应时间为12 h)。反应结束后,样品以700 μL乙酸乙酯萃取,有机层经无水碳酸钠干燥后进行气相色谱分析。
1.2.4 蛋白纯化及酶活检测
将收集的新鲜细胞悬浮于10 mL缓冲液(20 mmol/L Tris-HCl,pH 8.0) 中,进行超声破碎。CHMO及其突变体因纯化非常困难,以细胞破碎液直接进行酶活检测;PAMO及其突变体蛋白进行亲和层析的进一步纯化,镍柱的纯化步骤为:10 000 r/min、4 ℃离心30 min;将上清加入已被平衡过的GE公司His标签蛋白纯化预装柱GE Healthcore Histrap FF Crude column (5 mL)上;以含有500 mmol/L NaCl 和25 mmol/L咪唑的Tris-HCl冲洗杂蛋白;以含有500 mmol/L NaCl和200 mmol/L咪唑的Tris-HCl冲洗得到目的蛋白。镍柱纯化之后,将目的蛋白进行过夜透析,最后在441 nm下测其吸光值,以此数值作为突变体蛋白的相对浓度。酶活检测实验中,底物均以20 mmol/L终浓度溶解于乙腈中,NADPH以2 mmol/L终浓度溶于Tris-HCl缓冲液(pH 8.0) 中。反应体系如下:总体积200 μL,底物和NADPH终浓度分别为1 mmol/L和 100 μmol/L,酶用量各有不同,最后以Tris-HCl缓冲液 (pH 8.0) 补足200 μL总反应体系。将反应体系置于分光光度仪(Spectrophotometer Beckman Counter DU800) 下测340 nm下的光吸收,从其吸光度随反应时间的变化测定其反应初速度,进而计算其酶活[25]。PAMO及其突变蛋白的蛋白浓度以在441 nm下的吸光值度量[26],而CHMO及其突变蛋白浓度以Bradford蛋白测定法测定[27]。
2 结果与分析
2.1 BVMO基因序列与晶体结构分析
为了探索BVMO的构效关系,我们以NCBI以及PDB等数据库中BVMO的相关信息为基础,对BVMO进行了生物信息学分析和晶体结构分析,如图1所示。BVMO晶体结构主要由FAD结构域和NADPH结构域组成,两者之间存在特殊的Hinge结构。此结构包括两条链,且在晶体结构中恰好与BVMO的底物结合口袋在空间上相近。Hinge结构的序列比对结果中,两条链中的短链 (HingeⅡ) 保守性非常高而另外一条长链 (HingeⅠ) 保守性非常低。HingeⅠ在CHMO与底物结合的晶体结构中是缺失的,表明这一段序列在催化过程中柔性比较大,形态难以在晶体中固定,推测其在底物识别和催化氧化过程中扮演着重要的角色。本研究以CHMO为研究模型,并选取了如图2所示的各种酮类 (包括脂肪酮、环戊酮和环己酮类脂环酮等) 作为底物,从底物特异性、催化活性和立体选择性等多个方面探索突变对BVMO催化行为的影响,试图揭示非保守HingeⅠ的功能。

图1 BVMO晶体结构分析以及Hinge结构序列比对
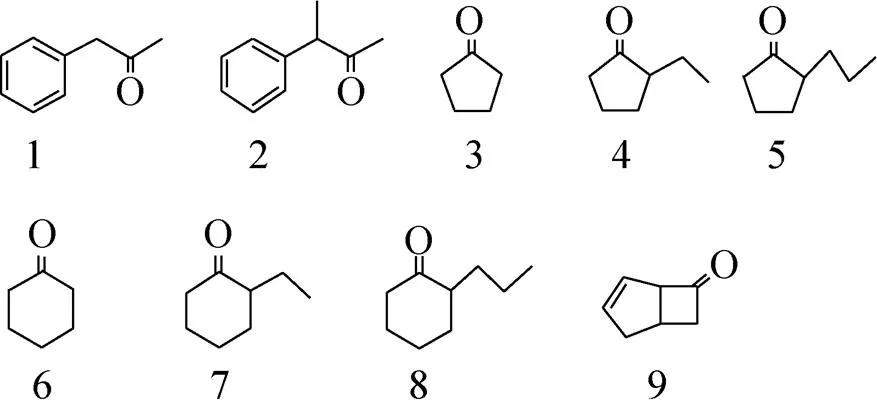
图2 本研究中涉及的主要底物
2.2 HingeⅠ功能检测——整体水平
为了从整体水平上考察HingeⅠ结构的功能,采用改进的PCR方法组合其他分子克隆手段得到了两个同源序列替换的突变体,CHMOPAMO和CHMOSTMO,它们是以CHMO为母体链的HingeⅠ位置分别被替换成PAMO和STMO的HingeⅠ的重组CHMO序列。将野生型CHMO与重组型进行全细胞水平的SDS-PAGE蛋白电泳,检测到三者的表达水平相似 (图3),证明HingeⅠ的整段替换并没有影响其表达水平。将它们分别对各种底物进行全细胞催化,并用气相色谱检测产物生成情况 (表1)。
对于CHMOSTMO来说,细胞基本上完全失活,只有底物9有极少量的产物生成。同源替换突变体CHMOPAMO则对简单脂肪环酮和底物9有较差的活性。野生型CHMO对这些底物有很高的催化活性。同时,那些野生型催化活性较低的底物则完全不能被CHMOPAMO所转化。可以看到,Hinge I序列的同源替换极大地损害了CHMO的催化活性,证明完整的Hinge I序列对CHMO的催化功能至关重要。
另外,CHMOPAMO突变体进行活性测试的底物中,不仅包括CHMO的活性底物,还包括PAMO的活性底物。PAMO HingeⅠ替换到CHMO HingeⅠ中,不但剥夺了CHMO本身的活性能力,而且也并没有赋予它任何类似PAMO的活性能力。说明不管对于CHMO还是PAMO,HingeⅠ结构同蛋白分子的其他部分都是协调合作共同起到相应的底物识别和催化作用的,“拆分嫁接”的后果就是两者整体性都被破坏,从而使酶失去催化功能。

图3 同源序列替换突变体表达水平检测

表1 全细胞催化同源序列替换突变体CHMOPAMO和CHMOSTMO
K: is abbreviated for ketone. +++: represents 50%−100% conversion; ++: 10%−50% conversion; +: 0−10% conversion; -: no conversion. Biotransformation was conducted at 32 ℃, lasted for 12 h.
2.3 Hinge I功能检测——个体水平
通过定点突变得到了10个CHMO Hinge I序列的丙氨酸扫描突变体,分别是:Leu143Ala、Leu144Ala、Ser145Ala、Pro147Ala、Asn148Ala、Leu149Ala、Pro150Ala、Leu151Ala、Ile152Ala和Lys153Ala。其中146位本身是丙氨酸,故而不在突变之列。
2.3.1 丙氨酸扫描突变体蛋白表达水平检测
所有的丙氨酸扫描突变体都用SDS-PAGE电泳的方法进行了蛋白表达水平的检测。与同源系列替换的突变体情况类似,所有的丙氨酸扫描突变体几乎都和CHMO野生型一样能大量表达,突变压力并没有给蛋白表达造成显著性影响。后文中我们得到的全细胞催化、气相检测和手性拆分中突变酶相对于野生型的改变并没有蛋白表达量的影响,其性质改变的原因源于酶本身的变化。
2.3.2 丙氨酸扫描突变体的活性检测
同样是全细胞催化、气相检测的手段鉴定各突变体的底物谱,得到表2。大多数突变体的活性都有部分下降,有些突变体活性改变较少,而有些突变体例如L144A的催化能力被大幅度削弱,特别是它与苯基丙酮的反应,在长达12 h的转化时间下转化率仍然为零。P147A、P150A和L151A则是活性较高的一类突变体,特别是在转化2位取代环己酮时,表现出比野生型更高的活性。
为了得到更为精确可靠的底物谱,还采用了细胞破碎、酶活测定的方法进一步检测突变酶的反应活性。由于CHMO纯化过程中损耗非常大,纯化过程困难,并且我们已经证明了野生型CHMO和突变型之间表达水平差异不大,所以以细胞破碎液 (粗酶) 为检测样品来比较野生型与突变型之间的酶活差异是合理的。将野生型CHMO和10个突变体的细胞破碎液分别与底物混合,并加入反应必需的辅基NADPH,反应体系放到紫外分光光度仪下检测25 ℃和 340 nm波长下的光吸收变化,从吸光值对时间的比率中可以得出反应初速率,再通过反应体系中的酶浓度即可得到能反映酶的催化能力的动力学指标——酶的比活。数据结果用图4的坐标轴方式清晰地展示出来。野生型CHMO在催化大部分底物中仍然是活性最高的一个,除了在催化2位取代环己酮时P147A和N151A两种突变体有微弱的优势。L144A在所有纵轴中都是排行最低的,证明其对比于野生型和其他突变体,在所有底物上都表现出最差的催化活性。图中K153A也是一个活性较差的突变体,尽管它在全细胞催化活性检测时并没有突出的表现。其他位于轴线中部的突变体比野生型CHMO催化能力差,但仍维持着中等的活性。
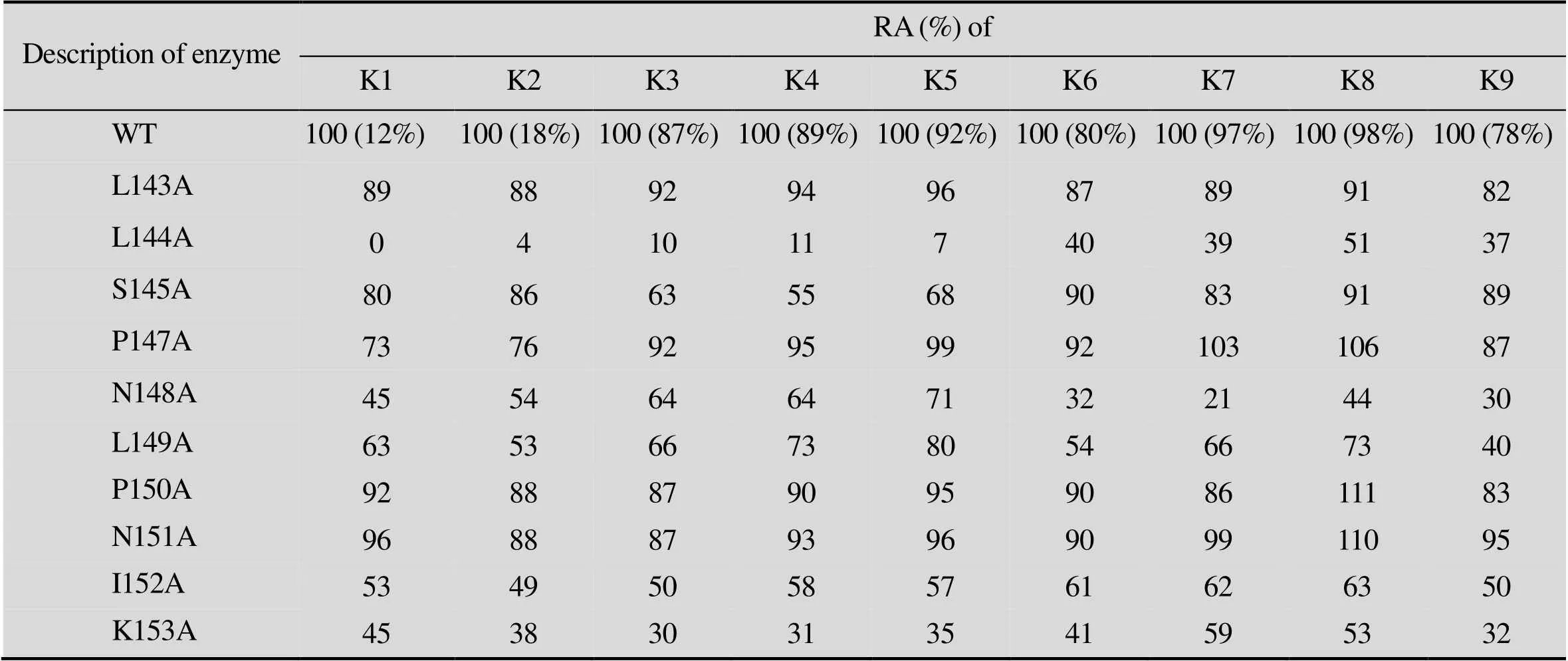
表2 全细胞催化检测CHMO丙氨酸扫描突变体的活性
K: is abbreviated for “ketone”; RA: relative activity. For a fair comparison, all incubations were for 12 h and relative activities are expressed as the rate of conversion normalized to that obtained with wild-type CHMO. Absolute conversion values for the wild-type CHMO are given in parentheses.

图4 酶活测定检测CHMO丙氨酸扫描突变体活性
图4的实验结果与表2基本上一致,除了K153A外,只有L143A在全细胞表达中产率较高而在细胞破碎液酶活测定中活性相对较低。同样作为检测催化能力的手段,全细胞催化着眼于整个活细胞的催化功能,不单单由酶本身的性质决定,更有酶的表达量以及细胞内NADPH浓度等各种其他因素的影响;而酶活检测则是细胞外相对精确的定量实验,更能体现酶本身的催化性质。
2.3.3 丙氨酸扫描突变体对映选择性检测
经过萃取得到CHMO及其突变体全细胞催化反应之后体系中的有机相,然后用手性气相色谱柱进行动力学拆分,得到一系列拆分数据,其中与野生型对映选择性有明显差异的部分如表3所示。L144A因其催化得到外消旋产物而再次成为表现突出的突变体,而在酶活检测中和L144A行为相似的K153A却有较高的化学选择性,特别是在催化2-丙基环戊酮时,甚至有比野生型更高的值 (值是度量催化剂立体选择性的指标,值越大代表选择性越好[28])。
双环[3,2,0]庚-2-烯-6-酮 (Bicyclo [3.2.0] hept-2-en-6-one,底物9) 是研究BVMO化学选择性的重要模式底物,因同时存在立体选择性和区域选择性,它在被BVMO催化时通常能产生各种构型构象的产物组合,如图5所示。有报道证明CHMO在催化底物9时有较高的立体选择性和较差的区域选择性[29],我们的结论与之一致。大部分CHMO突变体的行为也与野生型相似,反应生成(1S,5R) “正常”型酯和(1R,5S) “非正常”型酯。L144A在催化底物9时表现出较低的立体选择性,并且更倾向于生成“非正常”型酯,如表4所示。

表3 CHMO及其突变体的对映选择性检测
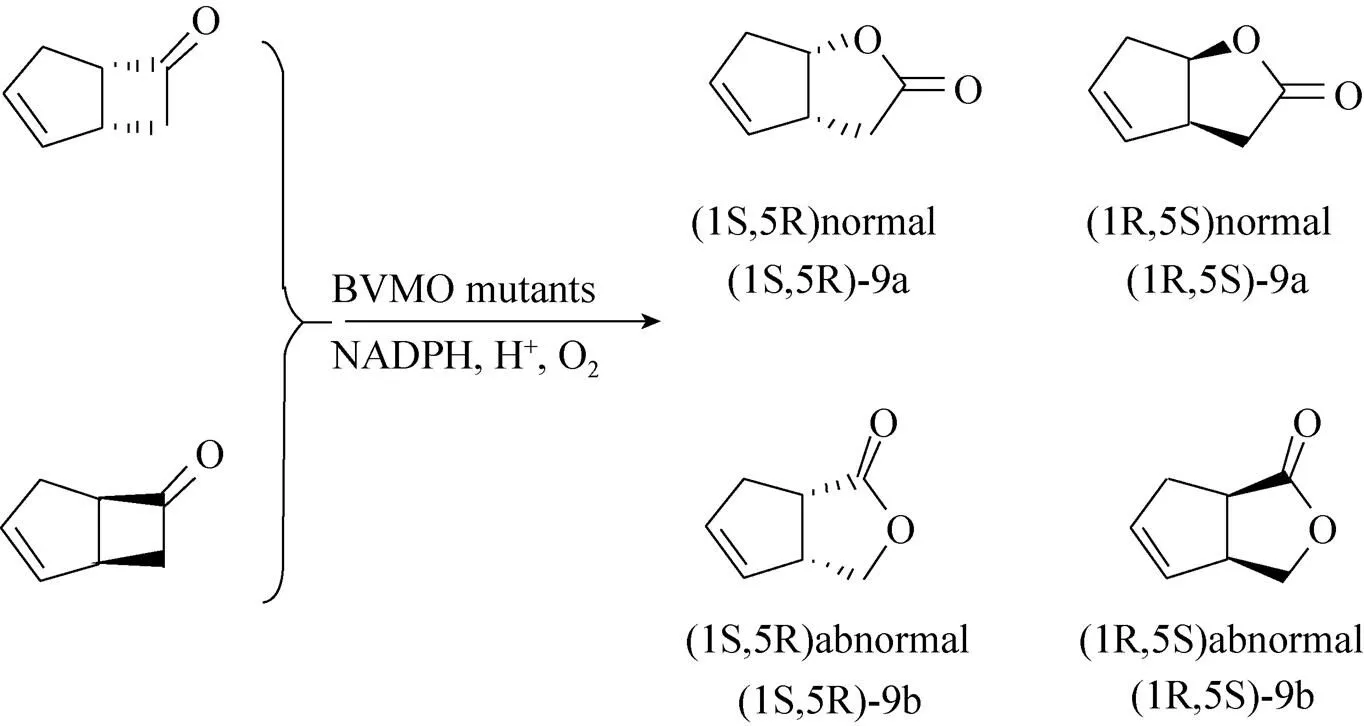
图5 BVMO催化底物9的可能结果

表4 CHMO及其突变体催化底物9的化学选择性
2.4 PAMO Hinge I的功能检测
从CHMO的实验结果中我们得到了一些显著影响其功能的重要位点,为了证明这些位点在其他BVMO中是否同样关键,将同样的研究方法运用于PAMO,得到的结论与CHMO非常一致。在催化PAMO经典底物如苯基丙酮、3-苯基-2-丁酮时,大部分突变体都显示出中等催化活性,与CHMO中L144A的对应突变体L153A则出现大幅度活性下降,立体选择性也下降。在催化非经典底物如2-丙基环戊酮、Bicyclo [3.2.0] hept-2-en-6-one (底物9) 时,大部分突变体催化活性呈现出和野生型一样的低催化活性,而L153A则活性相对较高。另外PAMO中还有非常值得注意的一个位点是Q152,其丙氨酸扫描突变体的活性和立体选择与L153A一样大幅度下降,对PAMO的影响程度不亚于K153。此外,CHMO中的对应位点突变体L143A在活性和选择性上也对CHMO有较大负面影响。我们还用SDS-PAGE检测了突变体的蛋白表达情况,发现其蛋白表达与野生型PAMO基本一致。
以上发现都说明Hinge I的重要位点在BVMO中很有可能具有普遍的影响力,为研究新型BVMO提供了宝贵的借鉴,同时给BVMO的定向改造和进化提供了新的突破口。
3 讨论
本研究以BVMO序列比对和晶体结构分析为基础,主要通过同源序列替换以及丙氨酸扫描突变的过程,得到了一系列BVMO突变酶;然后以改进的分子克隆技术、色谱技术以及酶学性质研究技术等为手段获得大量关于BVMO突变酶的底物选择性、反应活性以及对映选择性的实验数据。我们发现这些突变体在催化活性和化学选择性上相对于野生型来说有诸多改变,这些实验证据使我们能从中获取更多关于BVMO重要的构效信息。
从整体水平上,Hinge I的多氨基酸替换突变直接导致了CHMO催化功能的几乎完全丧失。事实上,在通过多个基因同时突变的手段来研究BVMO酶功能的变化时,Dudek等[31]就提到过“累积假说”的概念,即多个位点同时突变对酶功能的影响是由每个位点突变所产生的效应累积而成的。同源序列替换导致的活性完全丧失,表明了在此11个位点中,必然有能严重影响CHMO活性的位点存在,这也从侧面说明了本课题最开始的设想——HingeⅠ结构的重要性是符合事实的。当然,长达11个氨基酸的替换使酶活丧失,还有一个可能是其蛋白结构发生了重大改变从而无法进行任何催化造成的,但是我们替换进去的Hinge I序列是BVMO同源序列,同样能起到支持蛋白结构的作用。故而其酶活丧失最有可能的原因就是其中的某些非保守氨基酸残基在催化作用中起着关键性的作用。
从个体水平上,Hinge I丙氨酸扫描突变体表现各异,大多数维持着部分活性,少数活性与野生型相似,极少数活性得到了加强,比如PAMO L153A催化2-丙基环戊酮以及CHMO P147A催化2位取代环己酮,这或许可以成为我们拓展或者改变BVMO底物谱、定向改造BVMO的一个新的突破口。还有一类就是几乎完全丧失活性的突变,例如CHMO L144A。CHMO L144A是所有突变体中活性最低的一个,也是所有突变体中立体选择性最差的一个。我们知道“克利己” (Criegee) 中间体是BVMO催化过程中的一个关键形态,酶结构域的相对运动与酶和底物识别、结合密切相关,也即“Criegee”中间体的形成密切相关。CHMO与底物结合的晶体结构模型中,也可以看作是“Criegee”中间体模型中,Hinge I是一段在催化过程中有着非常频繁结构变动的区域。这段区域影响着整个酶的形态变化以及催化行为。作为这段区域的第一个氨基酸残基位点,L144或许在整条链的摆动运动中有着重要地位,直接影响着整个BVMO的催化过程。丙氨酸和CHMO此位置上原始的亮氨酸结构上差异并不大,只有一个单位的CH2的差异,但是却能使酶产生如此大的变化——酶活大大下降,选择性几乎完全丧失。可见这一位点在CHMO中的关键作用。CHMO L144位点有着很高的研究价值和改造潜能。
对比其他的BVMO,CHMO L144位点上的亮氨酸非常保守。我们将其对应位点PAMO的L153位点突变成丙氨酸后,也发现有催化能力的剧烈下降。研究发现此位点同Q152位点一样,处于底物结合区域的5 Å范围之内[31],它的突变将有很大可能性对PAMO产生影响。CHMO L144位点和PAMO L153位点的相似性表明,这一位点的重要性在所有的BVMO中很可能具有普遍性。
在CHMO突变体的催化选择性检测中,我们发现K153A突变体的活性有所降低,但是在立体选择性上却比野生型CHMO更加优秀,这是一个令人惊喜的正向进化。值得注意的是,在大多数BVMO的此位点上,都是脯氨酸 (Proline),包括PAMO在内。可见CHMO这个位点的赖氨酸对其立体选择性有着十分重要的影响。赖氨酸的侧链长度比脯氨酸长很多,这个位点很可能是通过侧链的构造影响底物结合口袋的形态,进而影响产物构型形成的。
综上所述,作为连接两个结构域的在催化过程中有着明显摆动的Hinge I结构对BVMO的催化功能有着关键作用,其中最开始的两个氨基酸以及最后一个氨基酸影响作用最为明显,是构成整条链的重要性的主要原因。对BVMO家族中非保守Hinge I结构的研究结果有助于精确描述BVMO底物结合口袋,进一步细化催化图景并对未来BVMO理性进化和定向改造研究奠定基础。
[1] Renz M, Meunier B. 100 years of Baeyer-Villiger oxidations. Eur J Org Chem, 1999, 1999(4): 737–750.
[2]Baeyer A, Villiger V. Einwirkung des Caro’schen reagens auf ketone. Ber Dtsch Chem Ges, 1899, 32(3): 3625–3633.
[3]Strukul G. Transition metal catalysis in the Baeyer-Villiger oxidation of ketones. Angew Chem Int Ed, 1998, 36(13/14): 1198–1209.
[4]Donoghue NA, Norris DB, Trudgill PW. The purification andproperties of cyclohexanone oxygenase fromCL1 andNCIB 9871. Eur J Biochem, 1976, 63(1): 175–192.
[5]Morii S, Sawamoto S, Yamauchi Y, et al. Steroid monooxygenase of: sequencing of the genomic DNA, and hyperexpression, purification, and characterization of the recombinant enzyme. J Biochem, 1999, 126(3): 624–631.
[6]Kamerbeek NM, Moonen MJH, van der Ven JGM, et al. 4-Hydroxyacetophenone monooxygenase fromACB. Eur J Biochem, 2001, 268(9): 2547–2557.
[7]Iwaki H, Hasegawa Y, Wang S, et al. Cloning and characterization of a gene cluster involved in cyclopentanol metabolism insp. strain NCIMB 9872 and biotransformations effected by-expressed cyclopentanone 1,2-monooxygenase. Appl Environ Microbiol, 2002, 68(11): 5671–5684.
[8]Pazmiño DET, Dudek HM, Fraaije MW. Baeyer–Villiger monooxygenases: recent advances and future challenges. Curr Opin Chem Biol, 2010, 14(2): 138–144.
[9]Fraaije MW, Kamerbeek NM, van Berkel WJH, et al. Identification of a Baeyer-Villiger monooxygenase sequence motif. FEBS Lett, 2002, 518(1): 43–47.
[10]Torres Pazmiño DE, Dudek HM, Fraaije MW. Baeyer-Villiger monooxygenases: recent advances and future challenge. Curr Opin Chem Biol, 2010, 14(2): 138–44.
[11]Van Beilen JB, Mourlane F, Seeger MA, et al. Cloning of Baeyer-Villiger monooxygenases from,andusing polymerase chain reaction with highly degenerate primers. Environ Microbiol, 2003, 5(3): 174–182.
[12]Mascotti1 ML, Ayub MJ, Dudek H, et al. Cloning, overexpression and biocatalytic exploration of a novel Baeyer-Villiger mononoxygenase fromAf293. AMB Express, 2013, 3(1): 33.
[13]Kotani T, Yurimoto H, Kato N, et al. A novel acetone metabolism in a propane-utilizing bacteriumsp. Strain TY-5. J Bacteriol, 2007, 189(3): 886–893.
[14]Iwaki H, Wang S, Grosse S, et al.cyclopentadecanone monooxygenase displaying an uncommon spectrum of Baeyer-Villiger oxidations of cyclic ketones. Appl Environ Microbiol, 2006, 72(4): 2707–2720.
[15]Mihovilovic MD, Bianchi DA, Rudroff F. Accessing tetrahydrofuran-based natural products by microbial Baeyer-Villiger biooxidation. Chem Commun, 2006, 14(30): 3214–3216.
[16]Gibson M, Nur-e-alam M, Lipata F, et al. Characterization of kinetics and products of the Baeyer-Villiger oxygenase mtmoiv, the key enzyme of the biosynthetic pathway toward the natural product anticancer drug mithramycin from. J Am Chem Soc, 2005, 127(50): 17594–17595.
[17]Willand N, Dirié B, Carette X, et al. Synthetic EthR inhibitors boost antituberculous activity of ethionamide. Nat Med, 2009, 15(5): 537–544.
[18]Moonen MJH, Rietjens I, van Berkel WJH. 19F NMR study on the biological Baeyer–Villiger oxidation of acetophenones. J Ind Microbiol Biotechnol, 2001, 26(1/2): 35–42.
[19]Malito E, Alfieri A, Fraaije MW, et al. Crystal structure of a Baeyer-Villiger monooxygenase. Proc Natl Acad Sci USA, 2004, 101(36): 13157–13162.
[20]Mirza IA, Yachnin BJ, Wang S, et al. Crystal structures of cyclohexanone monooxygenase reveal complex domainvmovements and a sliding cofactor. J Am Chem Soc, 2009, 131(25): 8848–8854.
[21]Kamerbeek NM, Olsthoorn AJJ, Fraaije WM, et al. Substrate specificity and enantioselectivity of 4-hydroxyacetophenone monooxygenase. Appl Environ Microbiol, 2003, 69(1): 419–426.
[22]Beam MP, Bosserman MA, Noinaj N, et al. Crystal structure of Baeyer-Villiger monooxygenase MtmOIV, the key enzyme of the mithramycin biosynthetic pathway. Biochem, 2009, 48(21): 4476–4487.
[23]Yachnin BJ, Sprules T, McEvoy MB, et al. The substrate-bound crystal structure of a Baeyer-Villiger monooxygenase exhibits a Criegee-like conformation. J Am Chem Soc, 2012, 134(18): 7788–7795.
[24]Hogrefe HH, Cline J, Youngblood GL, et al. Creating randomized amino acid libraries with the QuikChange®multisite-directed mutagenesis kit.Biotechniques, 2002, 33(5): 1158–1165.
[25]Suske WA, van Berkel WJH, Kohler HPE. Catalytic mechanism of 2-hydroxybiphenyl 3-monooxygenase, a flavoprotein fromHBP1. J Biol Chem, 1999, 274(47): 33355–33365.
[26]Reetz MT, Wu S. Laboratory evolution of robust and enantioselective Baeyer-Villiger monooxygenases for asymmetric catalysis. J Am Chem Soc, 131(42): 15424–15432.
[27]Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem, 1976, 72(1): 248–254.
[28]Straathof, AJJ, Jongejan JA. The enantiomeric ratio: origin, determination and prediction. Enzyme Microb Technol, 1997, 21(8): 559–571.
[29]Alphand V, Furstoss R. Microbiological transformations. 22. Microbiologically mediated Baeyer-Villiger reactions: a unique route to several bicyclic. gamma-lactones in high enantiomeric purity. J Org Chem, 1992, 57(4): 1306–1309.
[30]Szolkowy C, Eltis LD, Bruce NC, et al. Insights into sequence–activity relationships amongst Baeyer-Villiger monooxygenases as revealed by the intragenomic complement of enzymes fromRHA1. Chem Bio Chem, 2009, 10(7): 1208–1217.
[31]Dudek HM, de Gonzalo G, Pazmiño DET, et al. Mapping the substrate binding site of phenylacetone monooxygenase fromby mutational analysis. Appl Environ Microbiol, 2011, 77(16): 5730–5738.
(本文责编 陈宏宇)
Nonconserved hinge in Baeyer-Villiger monooxygenase affects catalytic activity and stereoselectivity
Qiuling Liang1,2, and Sheng Wu1
1,,,100101,2,100049,
Baeyer-Villiger monooxygenases (BVMOs) are important biocatalysts to synthesize a series of valuable esters and lactones. Based on protein sequence alignment and crystal structure analysis, a nonconserved hinge which linked NADPH domain and FAD domain was speculated to play an important role in substrate recognition and catalytic oxidation process. Cyclohexanone monooxygenase (CHMO) was selected as a model. Mutants obtained by homologous replacement of the whole hinge almost completely lost its original catalytic activity, demonstrating that the overall hinge structure was of great importance. Some significant sites were identified to greatly affect the catalytic activity and stereoselectivity by alanine scanning mutagenesis, accompanied by enzyme activity assessments and chiral kinetic resolutions. Altering K153 decreased the activity of the enzyme but enhanced the stereoselectivity. Changing L143 site reduced stereoselectivity but had little effect on enzyme activity. Mutation at L144 site dramatically weakened both activity and stereoselectivity. Subsequently, these corresponding sites in phenylacetone monooxygenase were also illustrated to follow a similar rule, revealing a universal importance of these sites in the BVMO family. These results expanded our understanding of the structure-activity relationship of these enzymes and provided more proofs for future directed evolution of BVMOs.
Baeyer-Villiger monooxygenase, cyclohexanone monooxygenase, function, homologous replacement, alanine scanning mutagenesis
May 27, 2014; Accepted: June 4, 2014
Sheng Wu. Tel: +86-10-62628482; Fax: +86-10-64807429; E-mail: shengwu@im.ac.cn
Supported by:National Natural Science Foundation of China (No. 31070718), Knowledge Innovation Program of the Chinese Academy of Sciences (No. KSCX2-EW-J-6).
国家自然科学基金 (No. 31070718),中国科学院微生物所基础前沿研究项目 (No. KSCX2-EW-J-6) 资助。
猜你喜欢
中华养生保健(2020年3期)2020-11-16
科学(2020年2期)2020-08-24
广东医科大学学报(2020年6期)2020-02-06
生命科学研究(2018年1期)2018-05-29
实验与检验医学(2017年6期)2018-01-20
上海农业学报(2017年3期)2017-04-10
安徽医科大学学报(2016年12期)2017-01-15
中外医疗(2015年11期)2016-01-04
天津医科大学学报(2015年2期)2015-12-22
电源技术(2015年10期)2015-08-01
