
几种新型植物基因表达载体的构建方法
2015-07-19 13:08张阳璞杨淑慎
生物工程学报 2015年3期
张阳璞,杨淑慎
几种新型植物基因表达载体的构建方法
张阳璞,杨淑慎
西北农林科技大学生命科学学院,陕西杨凌 712100

张阳璞, 杨淑慎. 几种新型植物基因表达载体的构建方法. 生物工程学报, 2015, 31(3): 311–327.Zhang YP, Yang SS. Methods for construction of transgenic plant expression vector: a review. Chin J Biotech, 2015, 31(3): 311–327.
利用基因工程技术手段研究基因功能过程中,构建基因表达载体处于转基因植物的主导地位,采用合适的构建方法会使实验效果事半功倍。植物基因表达载体的构建方法除了传统构建法、Gateway技术、三段T-DNA法、一步克隆法等,还有近年来出现的几种新型的载体构建方法:基于竞争性连接原理快速构建小片段基因表达载体;MicroRNA前体PCR置换法适用于构建小分子RNA表达载体;重组融合PCR法特别适用于插入片段中含有较多限制性酶切位点的载体构建;利用In-Fusion试剂盒可以将任何目的片段插入一个线性化载体的某个区域;构建多片段复杂载体可采用不依赖序列和连接的克隆方法(Sequence and ligation-independent cloning, SLIC) 法;Gibson等温拼接法;Golden Gate拼接法。本文将在总结分析前人工作的基础上,结合自己工作的体会和经验分析这7种新方法的特点,期望通过这几种新的方法给植物基因工程表达载体的构建提供新的思路。
MicroRNA前体PCR置换法,In-Fusion试剂盒法,重组融合PCR法,Gibson等温拼接法,Golden Gate拼接法
基因克隆、载体构建是植物功能基因组研究中的常规步骤[1]。而载体构建是基因工程和分子生物学研究中常用的基础技术。随着植物基因工程技术的发展,适合于不同研究目的各种载体系统应运而生,其中在转基因植物中最常用的是质粒载体[2]。传统的载体构建方法在进行构建多片段拼接的复杂载体时,需要精心选择酶切位点,有时还需要构建多个中间载体,操作比较麻烦,费时费力,因此寻找简单、高效、快捷的载体构建方法具有重要的现实意义。从1969年Arber等[3]发现了限制性内切酶,载体的构建方法逐步发展,从传统构建方法到三段T-DNA、Gateway等技术延伸出了许多新的载体构建方法。本文结合自己的实验工作选择介绍了近年来其中几种新型的具有代表性的植物表达载体构建的方法,对其应用的方向、优缺点作出了评估,期望给植物基因工程表达载体的构建提供新的思路。
1 载体构建方法
1.1 快速构建小片段基因表达载体
基因产物克隆的方法有很多种,如共环消解法、T4 DNA聚合酶回切产生粘端、外切核酸酶Ⅲ回切产生粘末端、PCR产物非依赖连接克隆、TA克隆等,这些方法原理不一,应用的方向也不相同,但都不适合小片段基因的克隆及其载体的构建[4]。传统构建方法构建载体时需要PCR扩增,用2个不同的限制性内切酶酶切PCR产物和载体,酶切后进行胶回收等,步骤繁琐、连接成功率低,任何一个步骤出现问题都会导致最终实验失败。因此,提高酶切和连接的效率是提高实验成功率的关键。
通常在实际的实验中我们会将酶切后的片段进行回收浓缩,而且使用较小的连接体系,这种方法就利用了竞争性连接的特点,来源于化学中的有效碰撞原理,连接反应也是一个化学反应,当在进行连接反应时,单位时间内分子数目多的目的片段分子更加容易与载体分子接触,换言之,有效分子浓度更高,则更容易发生连接反应,同时能有效减少载体自连反应。
小片段基因载体构建时,小片段的PCR技术又比较困难,尤其是在酶切回收步骤,也常出现酶切后的粘性末端被降解的现象[4],因此需要采取办法避免这些常见问题。金磊等[4]提出的新方法是基于小片段基因寡聚核苷酸合成技术和竞争性连接原理。利用寡聚核苷酸合成的方法,省去了目的基因的酶切步骤和载体酶切后胶回收纯化步骤,解决了小片段PCR困难的问题;利用竞争性连接原理可以克服载体自连,同时提高连接效率。其应用该方法已经完成了4个小片段基因(67 bp) 表达载体的构建,连接成功率达到66.7%−100%。证明了该方法具有简单快速、节省试剂费用和连接效率高等特点 (具体流程见图1A)。
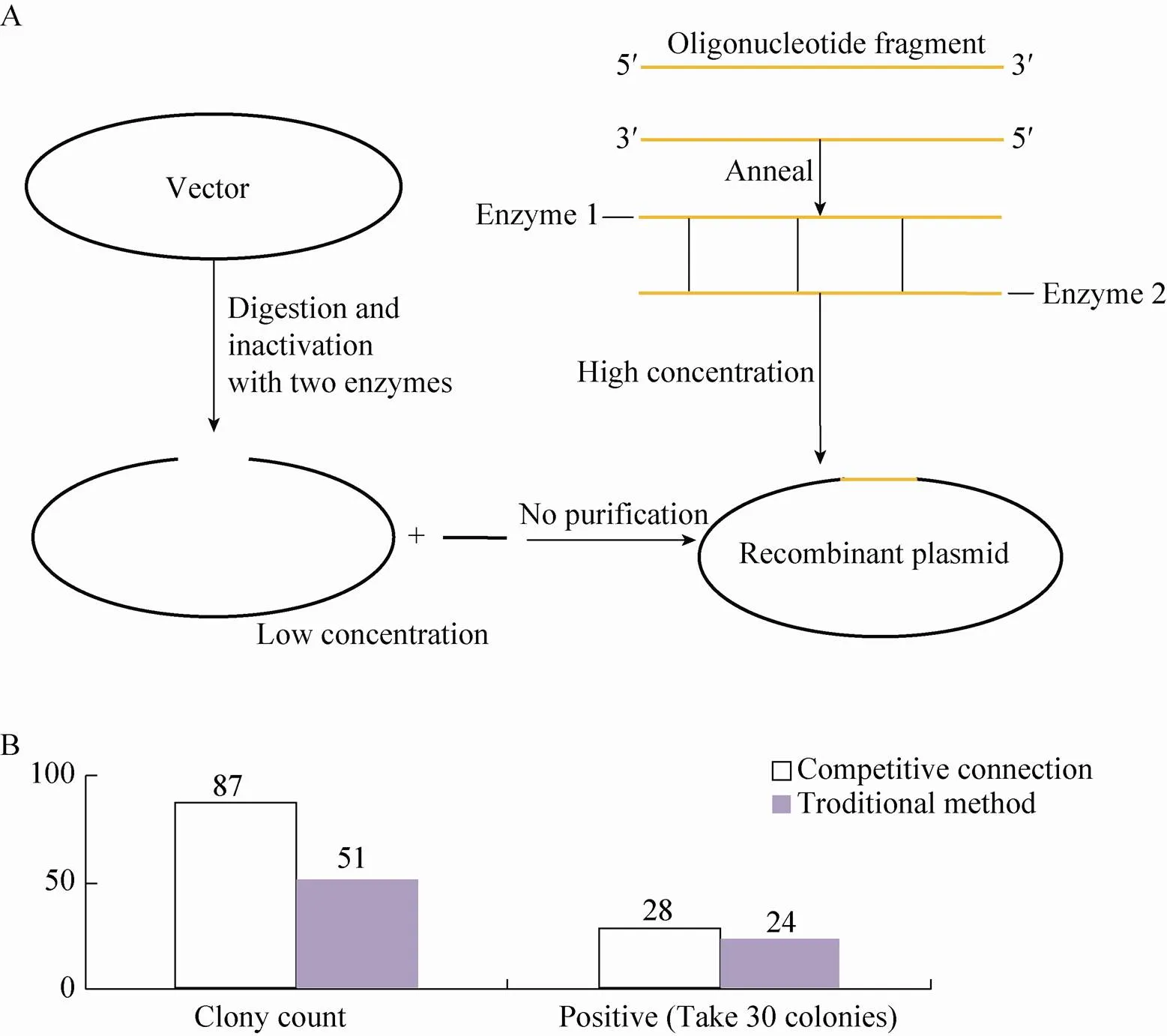
图1 小片段竞争性连接示意图(A)和两种方法的差异对比图(B)
虽然寡核苷酸可以通过化学方法合成[5-7],但是当寡核苷酸的长度超过100 nt (核苷酸) 时,随着长度的增加化学合成的质量急剧下降,而且成本会上升,也会增加回收纯化的难度[8-9],因此此方法在构建100 nt以上寡核苷酸片段时有一定的局限性。如何增加合成的寡核苷酸的长度也将成为此方法推广的关键所在。
在目的片段较长时也能利用到竞争性连接原理提高载体构建效率。笔者曾根据此原理成功构建了pTCK303-GAPDH (Glyceraldehyde-3- phosphate dehydrogenase,甘油醛-3-磷酸脱氢酶) 表达载体,根据单克隆菌落的数目和阳性菌落的数目对比 (图1B),反映了在转化的菌落数目上和阳性克隆的比率上有所提高。通常构建植物表达载体时,目的基因的长度一般比较大,根据上述方法的原理,同样可以通过增加酶切后目的基因片段的浓度来减少载体的自连反应,增加连接反应的效率。实验过程中增加酶切后目的基因浓度的方法有很多种,比如:扩增目的基因后使用DNA纯化回收试剂盒回收目的片段或者平行多次扩增目的基因后一起进行回收然后酶切等。
1.2 MicroRNA前体PCR置换法
自1999年Hamilton等[10]首次发现了长度为25 nt的RNA中间产物后,RNAi技术被广泛应用于植物基因功能鉴定和功能基因表达调控等各个领域。前人的研究中构建RNAi载体的方法主要有:传统的酶切连接法、Gateway技术、重叠延伸PCR法、LIC克隆法和Golden Gate克隆法[11]。
重叠延伸PCR技术(Gene splicing by overlap extension PCR,简称SOEPCR),于1989年由Horton等[12-13]建立,主要方法是采用具有互补末端的引物,使PCR产物形成了重叠链,从而在随后的扩增反应中通过重叠链的延伸,将不同来源的扩增片段重叠拼接起来。此技术利用PCR技术能够在体外进行有效的基因重组,而且不需要内切酶消化和连接酶处理,用这一技术可以很快获得其他依靠限制性内切酶消化的方法难以得到的产物[14],如Cao等[15]利用寡聚核苷酸合成技术和重叠延伸PCR技术合成了布氏柠檬酸杆菌植酸酶基因,并检测了其高效的表达。应用此方法还可以对目的基因进行小泛素相关基因的修饰,如Lu等[16-17]利用这种方法对HV1蛋白进行了泛素化修饰从而解决了此蛋白在大肠杆菌内外源表达易被降解的问题,以及泛素化修饰鸽子B淋巴细胞刺激因子() 增强了其在大肠杆菌内的可溶性表达。但是需要指出的是重叠延伸PCR技术在实际应用中,经常会受到引物自身序列的限制,例如,引物同 (异) 二聚体的产生导致的扩增效率低下,扩增片段中的重复序列导致产物突变等诸多问题[18]。
陈锐等[19]在研究中以成功构建携带预设小分子RNA的人工miRNA前体(Artificial pre- miRNA,apmiRNA) gso8-ap-miRNA为例介绍了一种新型的小分子RNA表达载体构建方法。此新方法利用了miRNA的单链前体(Pre-miRNAs) 具备特征性的发卡环二级结构,再利用分段重叠延伸PCR法置换成熟链及星号链为目的片段 (图2)。该方法为小分子RNA的功能研究提供了便捷有效的途径。
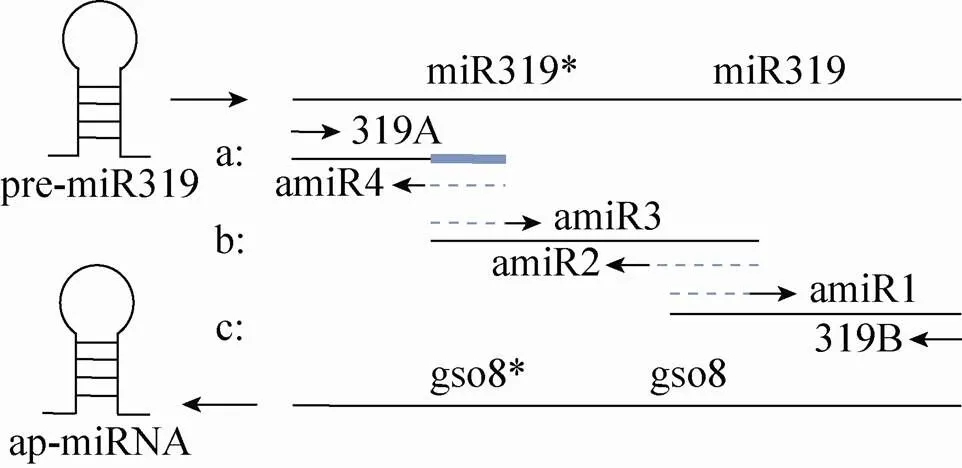
图2 置换PCR示意图[19]
笔者在构建基因的干扰载体时发现,虽然选取了需要沉默的基因中的部分片段作为构建干扰载体的目的基因,但是这些植物本身的基因并不含有特定的二级结构,这样依照此法构建人工miRNA前体时,可能不能更有效地表达外源小分子RNA。另外,此种方法理论基础是内源的miRNA成熟加工机制,但是目前一些miRNA代谢通路上的生成机制尚不清楚,这些都使得此方法在应用上具有一定的局限性[19]。在构建不含有特异的miRNA作为骨架的干扰载体时,可以使用In-Fusion试剂盒法将小干扰片段插入到载体的任意位置。
1.3 重组融合PCR法
基因的同源重组是噬菌体、细菌到真核生物都普遍存在的生物学现象[20]。广义的同源重组是指含有同源序列的DNA分子之间或分子之内的重新组合[21-23],同源重组严格依赖DNA分子之间的同源性,因此,原核生物的同源重组通常发生在DNA复制的过程中,而真核生物的同源重组则常见于细胞周期的S期之后,DNA的修复过程中也会发生同源重组的现象[24]。以同源重组技术为基础,通过构建突变或缺失的同源媒介基因载体并取代基因组中野生型的等位基因,进而研究目的基因与表型性状间的关系,是研究动物、植物、微生物基因功能的一种非常有用的遗传操作方法[25-26]。
常用的同源重组克隆的策略包括:T4 DNA聚合酶介导的同源重组,ExonucleaseⅢ介导的同源重组,RF克隆等[18]。这些传统构建方法由于要避免目的片段中已有限制性酶切位点,而不得不选择构建中间载体或者选择昂贵且酶切效率低的非常用限制性酶,经过多次连接转化,操作麻烦,费时费力,不但大量增加了载体构建的工作量,而且实验成功得不到保证。融合PCR技术在不经过酶切和连接的条件下,采用具有互补末端的引物将不同来源的扩增片段连接起来,为同源重组片段的构建提供了快速简捷的途径[27-28]。现有的融合PCR技术一般包括两步PCR反应,类似于重叠延伸PCR法[12-13]和一步克隆法[30]:1) 分别设计5′端带有互补序列的若干条特异性引物,分别进行各个片段的扩增;2) 随后在同体系加入各片段,以一对外侧引物进行融合片段的全长序列的扩增。
邝翡婷等[31]以水稻胚乳特异性荧光表达载体PGt1-mGFP-pSB130为例,介绍一种结合同源重组和融合PCR两种方法的重组融合PCR构建载体方法(图4),重组融合PCR法主要有以下优势:1) 只要求骨架载体上有一个特异性酶切位点,特别适用于插入片段中含有较多限制性酶切位点的载体构建;2) 可以引入定点突变,可便捷构建分析启动子功能等需引入定点突变的载体[32];3) 实现多基因的无缝融合构建多顺反子,在构建原核基因表达载体、叶绿体表达载体、线粒体表达载体方面有着明显优势[31]。
以此方法构建带有标记基因的表达载体,可以检测目的基因的表达或者进行亚细胞定位,利用该方法可将目的基因与标记基因利用单个酶切位点一步克隆连接到目的载体上。总之,重组融合PCR法可以将不同来源的几个目的片段仅通过一次重组转化克隆到目的载体上,大量减少中间载体的构建,提高工作效率和实验成功率。但是在应用过程中还存在很多方面的问题,如融合产物长度一般在40 kb以下、待融合片段的个数一般不超过3个、产物特异性差等[27],这也给其广泛应用带来了一定的限制。
1.4 In-Fusion试剂盒法
构建载体时需要通过连接酶连接完成,而且选择酶切位点时通常会被载体上独特的酶切位点所限制[33],因此能够摆脱酶切位点的限制,省略酶切与连接的步骤,并且能够在载体的任意位置上插入目的基因的序列是载体构建发展的新趋势。Gateway技术、一步克隆法等技术的出现大大简化了载体构建的步骤,但仍然没有解决酶切位点的限制等问题。
近年来的研究发现,利用In-Fusion试剂盒(In-FusionTMadvantage PCR cloning kit) 能够摆脱酶切位点的限制,利用这种方法可以对任何常用的载体进行修饰,用单酶切或者利用PCR扩增的方法将载体线性化,使之成为一个不依赖序列和连接反应高效的基因克隆体系[34],该方法利用重叠PCR技术在PCR引物的5'端增加一个与线性载体两端同源的15 bp的序列,由于序列的同源性,通过PCR程序可将目的基因插入到载体中,实现DNA重组[35-36]。这种方法操作简单,对目的基因和载体没有特殊的要求,可以将任何目的片段插入一个线性化载体的某个区域,不会产生反向插入的问题,没有知识产权限制[36-37],而且适用于在多宿主中的表达[34]。
利用In-Fusion试剂盒构建表达载体的基本原理与步骤 (图3A)。1) 质粒的线性化(单酶切或双酶切);2) 目的基因的In-Fusion改造;3) 经过In-Fusion改造目的基因片段与线性载体在In-Fusion酶的作用下发生重组,使目的基因连接到载体上;4) In-Fusion产物的转化与检测;5) 提取阳性克隆质粒进行PCR和酶切验证[40]。
刘超等[38]利用In-Fusion技术成功将基因亚克隆到真核细胞表达载体pCMV- IRES-DsRed上得到pCMV-SCN1A-IRES-DsRed表达载体;林朝贵等[39]以此方法成功构建了p-GCFU-Survivin慢病毒表达载体;韩凯等[40]以此方法成功构建了玉米L谷氨酰胺合成酶基因 () 表达载体pCUbi1390-Ubi- GS1-35S-EPSPS,证明了其简单、高效、快速、无需酶切连接和能够无限制、不定点插入的 特点。
In-Fusion法与重组融合PCR法的不同点在于In-Fusion法可以根据质粒上的任意酶切位点将目的片段分多次插入 (图3B),而重组融合PCR法是将不同来源片段通过融合之后一步克隆到载体上,因此利用In-Fusion的方法更加有利于构建插入多个小片段的干扰载体,而重组融合PCR法更加适用于构建需要添加标记基因的载体。利用In-Fusion试剂盒法可以减少连接、转化的次数,且重组效率高。相对于Gateway技术来说,In-Fusion法更简单、更快速,只要设计特殊的In-Fusion引物扩增目的片段, 30 min就可以转化,将目的基因高效地转入载体中,广泛适用于各种表达载体的构建[40]。但是,在一些转基因植物研究中需要构建由Ti质粒衍生而来的双元载体来提高转基因的效率。双元载体普遍较大,不适合使用PCR扩增的方法将其线性化,这也是此方法的一个限制性因素。Berrow等[34]利用这一技术进行高通量载体构建时发现克隆效率超过90%,明显高于Gateway重组技术(79%) 和传统的限制性酶切连接技术(87%)[41]。
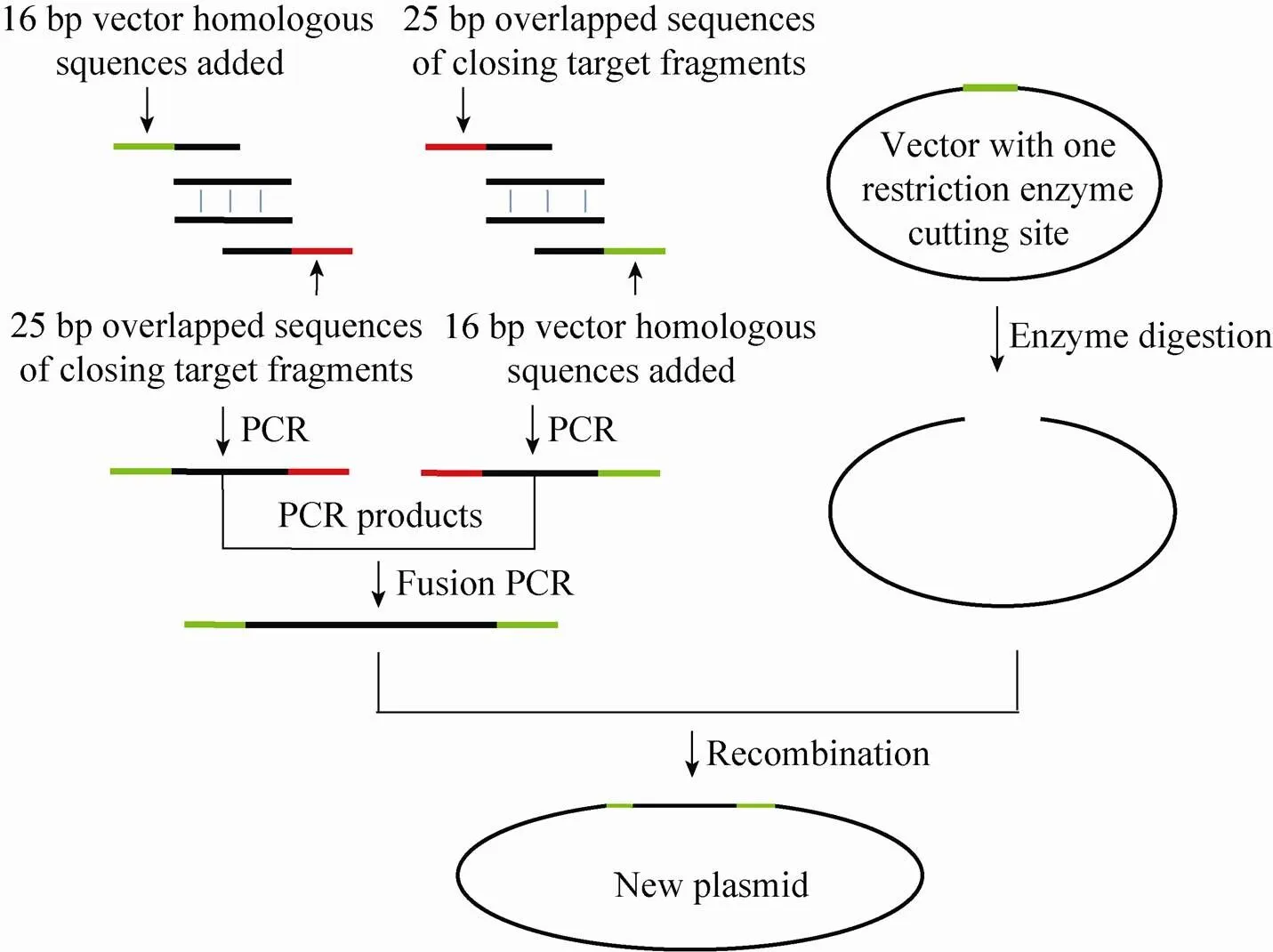
图3 重组融合PCR示意图
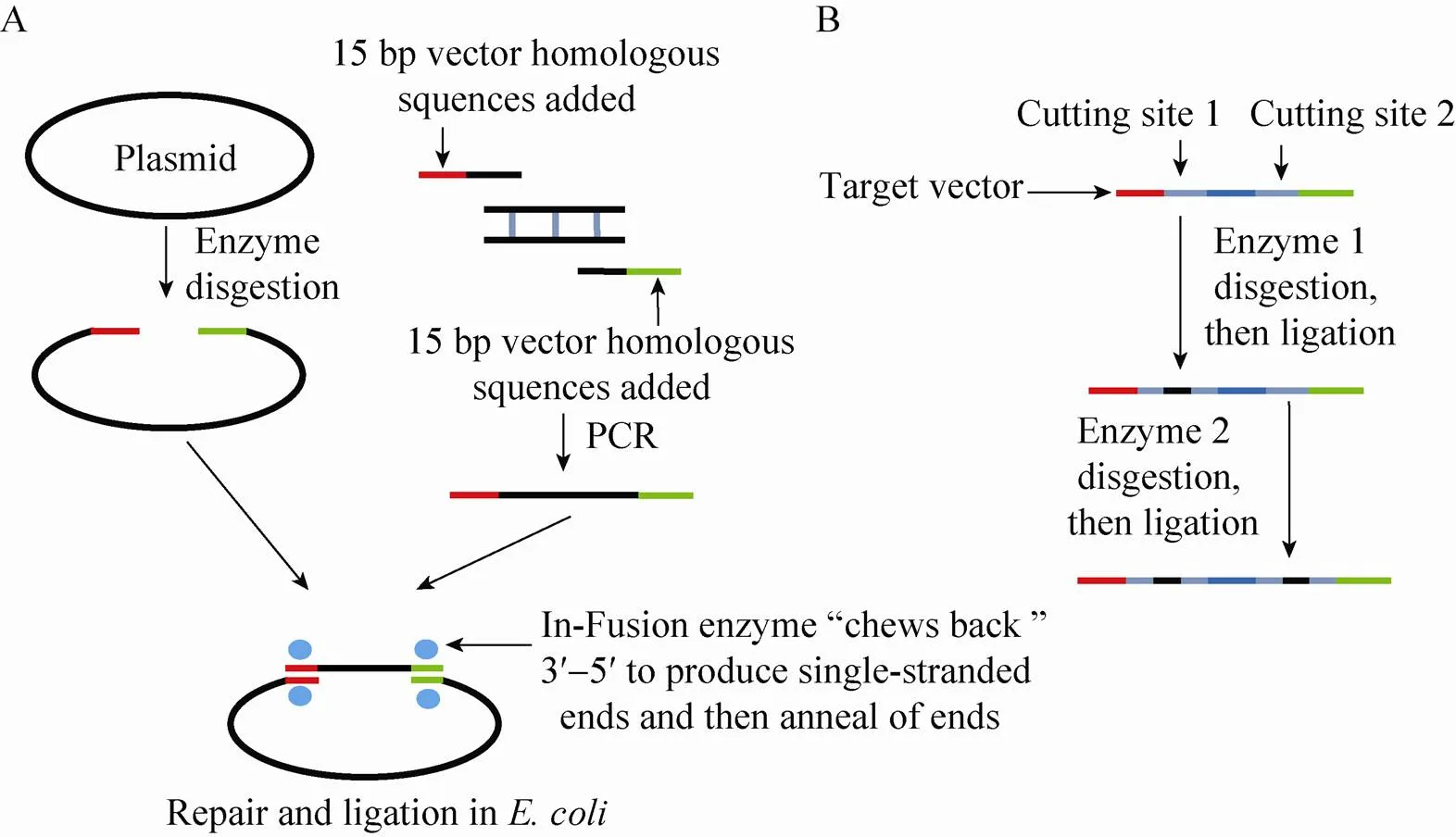
图4 In-Fusion试剂盒构建表达载体的步骤
1.5 SLIC法
在一些基因共表达的研究中,需要构建一些大型复杂载体,将若干个目的片段依次、连续连接到目标载体上,插入片段较多,酶切位点难以选择,通常的构建方法是使用平末端连接,但是平末端连接效率低且工作量与难度 较大。
Li和Elledge所建立的不依赖序列和连接的克隆方法(Sequence and ligation-independent cloning,SLIC)[42],把同源重组与单链退火结合起来,可以高效、定向地将任意序列的2个[43]或2个以上的DNA片段组装到一起,不需要连接反应即可完成体外的重组,避免目的基因序列中原有酶切位点对DNA重组的限制,极大地简化了DNA重组过程[44]。
采用SLIC法构建复杂载体的基本步骤 (图5):1) 采用PCR扩增使载体线性化;2) 在PCR的引物两端加上与载体末端同源的DNA序列 (20−30 bp),扩增目的基因;3) 用T4 DNA聚合酶处理目的基因和载体片段,使其5'末端形成突出单链;4) 在T4 DNA连接酶缓冲体系中退火,形成重组中间体;5) 转化大肠杆菌并筛选重组子[45]。
白帆等[45]在研究中证明了SLIC的重组是非常可靠的,能够在设计位点处发生精确重组。王广珺等[46]对此种方法进行了改进,将T4 DNA连接酶缓冲液改换成退火缓冲液,并使用相应的退火程序,可使重组效率提高至少10倍。Jeong等[47]对此方法进行了简化,称为一步SLIC (One-step SLIC),其改进是直接将扩增得到的目的基因与线性化载体混合后在室温下用T4 DNA聚合酶处理2.5 min,冰上孵育10 min直接转化大肠杆菌,并成功完成了4个片段的拼接。
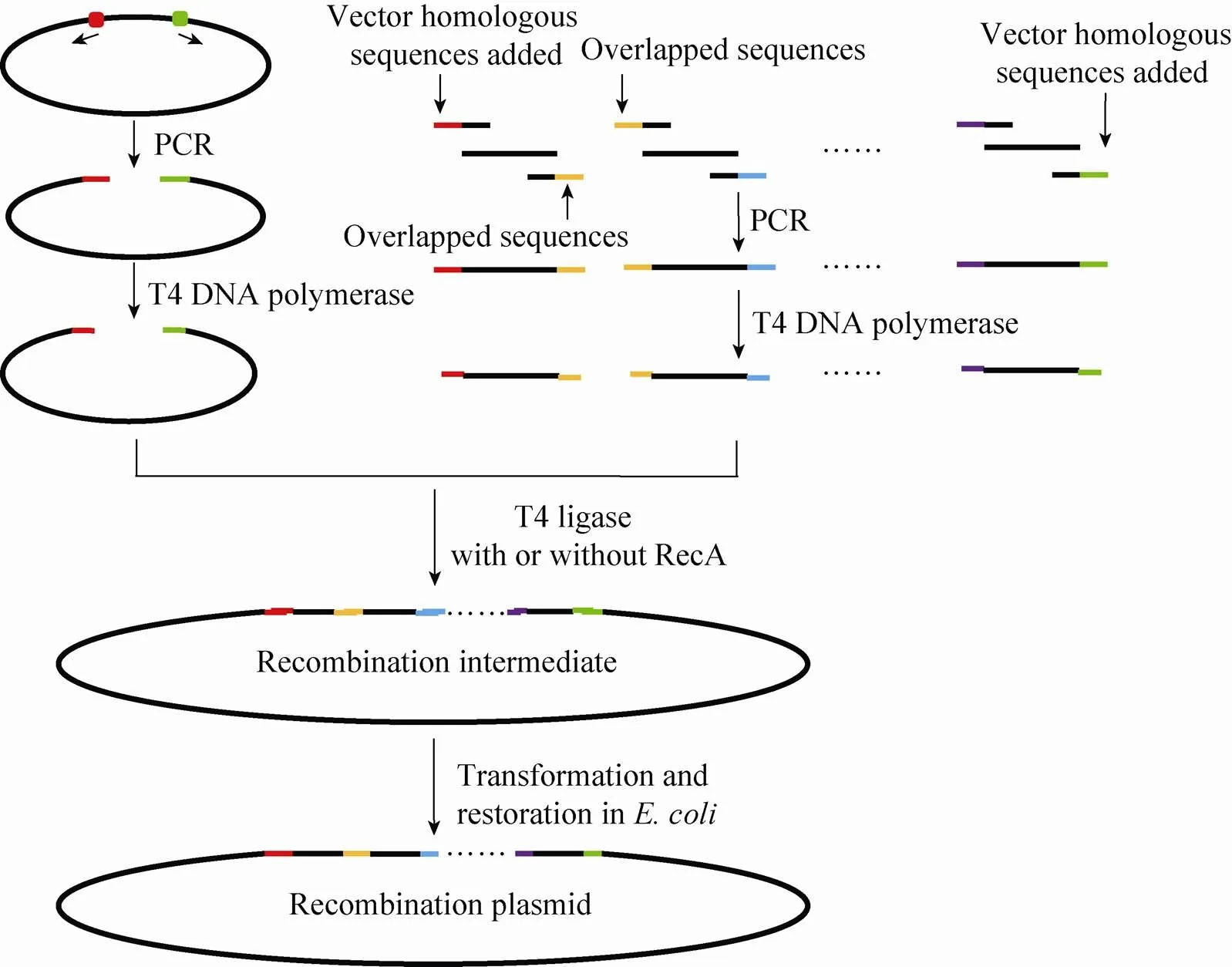
图5 SLIC法构建表达载体的步骤
采用SLIC法构建载体时,目的基因在2–8个之间效果较好,目的基因片段越多,筛选得到正确的重组子越困难,效率越低。在不超过5个基因片段连接的情况下,可以保证转化子100%为重组子,在10个基因片段连接的情况下,16%的转化子为正确的重组子[45]。值得注意的是,经过T4 DNA聚合酶处理后形成的ssDNA非常容易形成二级结构,从而影响DNA分子的互补、复性并降低重组效率[46]。另外,由于SLIC依赖于同源重组进行基因克隆,同源重组本身具有一定的随机性,因此可能产生不完整的重组子[43]。
1.6 Gibson等温拼接法
Gibson等温拼接法是Gibson等[48]报道的一种高效、快速的多基因片段拼接技术,由Gibson变温拼接法发展而来。与SLIC法相似,可以将任意序列的2个或2个以上的DNA片段组装到一起,适用于复杂载体的构建。不同的是,通过控制目的片段重叠序列的大小 (40−600 bp) 可以直接在体外合成得到较大的基因组序 列[49],大大简化了大型DNA分子合成的过程,展示了其广泛的应用前景。
采用Gibson法构建表达载体的步骤 (图6):1) 用两端带有与载体末端同源序列的引物PCR扩增目的基因;2) 将PCR产物加入3种酶 (Phusion或聚合酶、T5外切酶和DNA连接酶)[47-48]的混合缓冲液中;3) 混合液在50 ℃恒温孵育一段时间,时间由片段长短确定。高保真的DNA聚合酶具有3'→5'核酸外切酶的活性,扩增过程中可以将错配的碱基切掉,保证其准确性。除去扩增目的基因和载体片段的步骤,只需要一步等温反应就能完成片段的拼接,因此这一部分被Gibson命名为一步体外等温重组法 (One-step isothermalrecombination)[48]。
Sleight等[50]采用Gibson法成功组装了CMY (Cyan-Magenta-Yellow,青色-洋红色-黄色) 三基因表达载体,并且通过插入不同的启动子、核糖体结合位点和终止子,使得CFP、RFP和YFP三种荧光蛋白能够在外界环境调控下独立随机表达;Bartosiak-Jentys等[51]也用此种方法成功构建了由4片段基因组成的大肠杆菌-地芽胞杆菌sp.穿梭载体pUCG3.8,并发现其相对于原始的穿梭载体pUCG18转化热葡糖苷酶地芽胞杆菌的效率增加了2.8×105CFU/(μg DNA);Wagner等[52]使用Gibson法和传统的酶切-连接法都成功构建了恶性疟原虫载体,并证明Gibson法的效率明显高于传统的酶切-连接法。Gibson等成功在体外组装了多达583 kb的尿道支原体基因组序列[48]以及16.3 kb的鼠线粒体基因组序列[53],为基因组学的研究提供了新途径。
此种方法所用的3种酶均为实验室常见的3种酶,一步组装,简便、快速。在实际的操作中,核酸外切酶可能水解掉较短片段,因此此种方法不适合用于小片段基因的组装。Stefan等[54]在研究中发现在2个基因片段连接的情况下,可以保证转化子80%为正确的重组子,随着基因片段数目的增加,在8个基因片段连接的情况下,13%的转化子为正确的重组子,其效率比SLIC法略低。
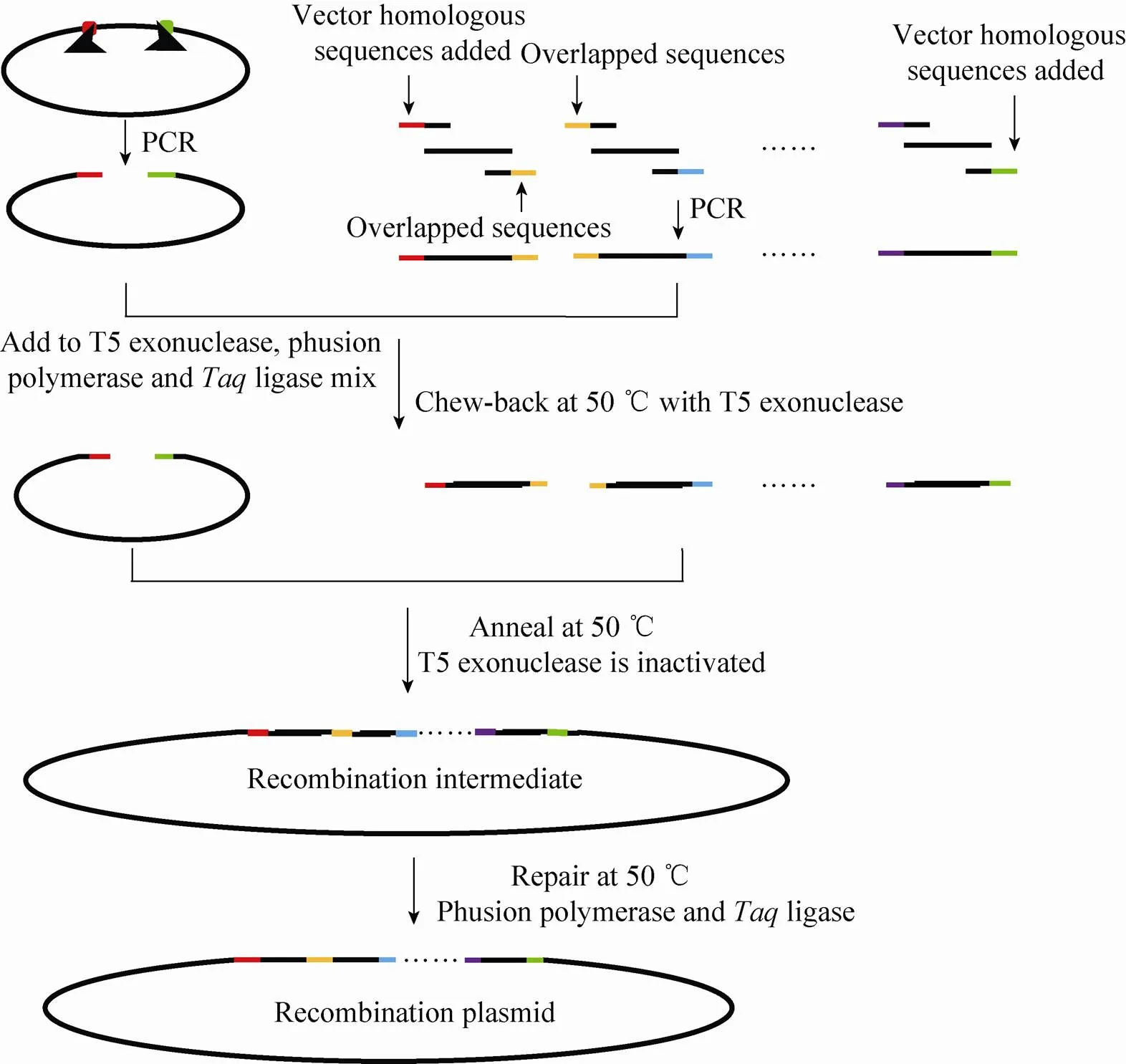
图6 Gibson等温拼接法构建表达载体的步骤
1.7 Golden Gate拼接法
在使用SLIC、Gibson法构建多片段复杂载体时,基因片段末端的ssDNA同源序列不能包含单链二级结构,且片段末端ssDNA同源序列应各不相同,否则会导致重组效率降低、片段的丢失和错排等现象。传统的Golden Gate克隆法构建复杂载体时不需要进行额外的PCR扩增和载体线性化的步骤,直接使用未经酶切的含有目的基因的载体在一个反应中完成多个序列的组装 (图7X)[55-58],可以对拥有重复的复杂二级结构的激活子进行拼接,如效应器TALEs[59]。
传统的Golden Gate克隆法又叫IIS克隆法,是Engler等报道的一种由IIS型限制性核酸内切酶介导的“无缝”连接的克隆策略。IIS型限制性内切酶有一个结合识别位点的域和一个专门切割DNA的功能域,常以二聚体形式作用于DNA序列,在识别位点之外切开DNA,产生具有黏性末端的DNA片段,再用连接酶将几个DNA片段按照既定的顺序拼接成不含酶识别位点的DNA片段[56-58]。Engler等[57]采用此法将9个片段拼接入目的载体,经过验证有90%的转化子为正确的重组子,证明了其在多片段连接上的高效性。
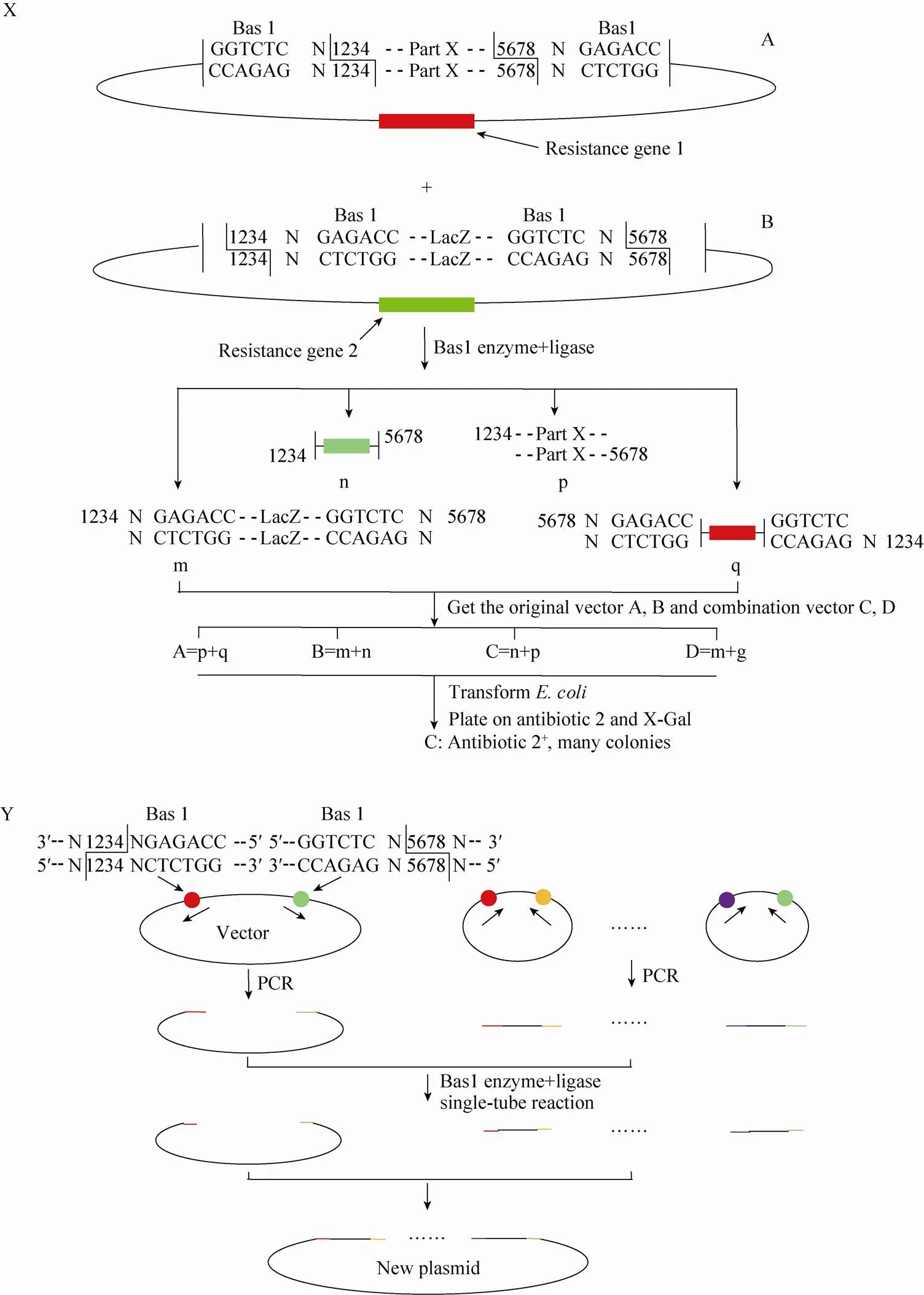
图7 Golden Gate克隆法 (X) 和Golden Gate AssemblyTM示意图 (以Bas1酶为例,Y)[54-56,63]
此种方法采用未经酶切的质粒作为材料,优点是可以解决PCR扩增所带来的碱基突变的问题,但是,它要求将所有待组装的片段必须克隆到Golden Gate质粒系统中,由于突出末端序列固定不变,大大降低了可以利用此法进行拼接的目的基因的种类,是此法在应用上的一个限制因素[60]。由于突出端序列的大小只有4 bp,那么在拼接反应中突出末端至少有一个,最好有两个碱基不同。当目的片段的数目大于10或者突出末端的GC含量高度偏向一个极端时,那么可能无法找到一组能够互相兼容的末端序列,那么在这种情况下就可以使用分级Golden Gate拼接法[61]。Weber等[62]和Werner等[63]将Golden Gate克隆法进一步延伸成为一个模块化的克隆策略 (Modular cloning,MoClo) 并以此方法分别完成了11个和17个独立转录片段的拼接。
Golden Gate拼接法 (Golden Gate AssemblyTMcloning kit) 与Engler的Golden Gate克隆法有所不同[55,64],采用PCR扩增的方法使载体线性化及得到目的基因片段,大大增加Golden Gate克隆法的应用范围。其基本步骤为 (图7Y):1) 实用软件对目的基因进行酶切位点分析;2) PCR扩增目的基因片段和线性化质粒载体;3) 将基因片段和线性化载体加入含有IIS型限制性内切酶和DNA连接酶的缓冲液中 (合适条件下) 进行反应。
2 讨论与展望
本文简要介绍了目前已经成熟的几种新型载体构建技术的原理、特点和应用,并对几种载体构建方法作了一个综合评价,表1中比较了7种不同载体构建方法的特点,指出了这些方法的应用方向和潜力。可以看出,由传统的构建方法到Gateway技术,再到本文列举的各种无酶连接的新方法,载体构建方法已经进入了无酶连接的新阶段。若构建小片段载体时可尝试基于寡核苷酸合成和竞争性连接原理的小片段基因载体构建的方法;如果在构建干扰载体时,若目标miRNA存在特异性的骨架,可以采用MicroRNA前体PCR置换法,如果没有特异性的骨架则使用In-Fusion试剂盒法更加简便;需要多片段连接的复杂载体或添加标记基因的载体可使用重组融合PCR法,而且利用此法可以引入突变位点,同时为研究基因功提供了新方法;若需要避开某些基因的功能区域或需要在不同位点快速插入不同的目的基因时,可以采用In-Fusion试剂盒法。若需要构建多片段复杂植物表达载体,根据序列多少、大小、是否需要保留特殊结构和经济等因素,在SLIC、Gibson等温拼接、Golden Gate拼接法中合理选择。随着植物基因工程技术的不断更新,各种植物载体构建技术正不断成熟,而其发展的必然趋势是更加简便、安全、通用、高效和经济。
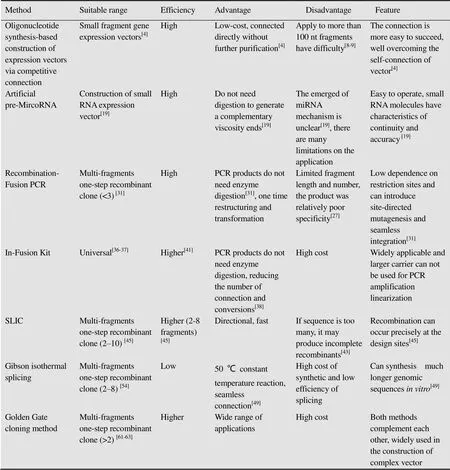
表1 几种主要构建方法的综合比较
[1] Baloglu MC, Battal A, Aydin G, et al. Vector consyruction strategies for transformation of wheat plant. J Anim Plant Sci, 2013, 23(3): 906–912.
[2] Lin CJ, Wei ZY, Cai QA. Approaches of constructing expressional vectors utilized in plant genetic transformation. Biotechnology, 2008, 18(5): 84–87 (in Chinese). 林春晶, 韦正乙, 蔡勤安. 几种植物转基因表达载体的构建方法. 生物技术, 2008, 18(5): 84–87.
[3] Kühnlein U, Linn S, Arber W. Host specificity of DNA produced by, XI.modification of phage FD replicative form. Proc Natl Acad Sci USA, 1969, 63(2): 556–562.
[4] Jin L, Zhao WX, Ma L. A novel method for construcying small fragment gene of expression vector. China Biotechnol, 2012, 32(6): 57–63 (in Chinese). 金磊, 赵文秀, 马岚. 新型小片段基因表达载体构建方法. 中国生物工程杂志, 2012, 32(6): 57–63.
[5] Zhao P, Zhang Z, Ke HG. Oligonucleotide-based targeted gene editing in.via the CRISPR/Cas9 system. Cell Res, 2014, 24(2): 247–250.
[6] Zhang PP, DingYY, Liao WT. A simple, universal, efficient PCR-based gene synthesis method: sequential OE-PCR gene synthesis. Gene, 2013, 524(2): 347–354.
[7] Su M, Wang J, Tang XJ. Photocaging strategy for functionalisation of oligonucleotides and its applications for oligonucleotide labelling and cyclisation.Chem-Eur J, 2012, 18(31): 9628–9637.
[8] Zhao SZ, Shen XQ. Investigation of biosynthesis method for long oligonucleotides. Acta Bot Bor-Occid Sin, 2010, 19(5): 47–51 (in Chinese).赵松子, 沈向群. 长链寡核苷酸生物合成方法的研究. 西北农业学报, 2010, 19(5): 47–51.
[9] Hoover DM, Lubkowski J. DNA works: an automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res, 2002, 30(10): E43.
[10] Hamilton AJ, Baulcombe DC. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science, 1999, 286(5441): 950–952.
[11] Yan P, Shen WT, Li XY. Progress in construction of hpRNA vector for plant RNAi. Biotech Bull, 2013, 29(9): 7–12 (in Chinese).言普, 沈文涛, 黎小瑛. 植物hpRNA干扰载体构建的研究进展. 生物技术通报, 2013, 29(9): 7–12.
[12] Liu LL, Tan Y, Liu LH. Splicing overlap extension by PCR: a convenient method to obtain recombinant gene. J Jilin University: Med Ed, 2004, 30(5): 713–716 (in Chinese). 刘玲丽, 谭岩, 刘力华. 获得重构基因的简捷方法—重叠延伸PCR. 吉林大学学报: 医学版, 2004, 30(5): 713–716.
[13] Warrens AN, Jones MD, Lechler RI, et al. Splicing by over lap extension by PCR using asymmetric amplification: animproved technique for the generation of hybridproteins of immunological interest. Gene, 1997, 186(1): 29–35.
[14] Xu F, Yao QH, Xiong AS. SOE PCR and its application in genetic engineering. Mol Plant Breeding, 2006, 4(5): 747–750 (in Chinese).徐芳, 姚泉洪, 熊爱生. 重叠延伸PCR技术及其在基因工程上的应用. 分子植物育种, 2006, 4(5): 747–750.
[15] Cao SS, Hu ZQ. A new method for gene synthesis and its high-level expression. J Microbiol Meth, 2009, 79(1): 106–110.
[16] Lu WG, Cai XT, Zhenghua Gu. Production and characterization of hirudin variant-1 by SUMO fusion technology in. Mol Biotechnol, 2013, 53(1): 41–48.
[17] Wuguang Lu, Peng Cao, Xueting Cai. Molecular cloning, expression, and bioactivity of dove B lymphocyte stimulator (). Vet Immunol Immunop, 2009, 128(4): 374–380.
[18] Dai Q, Tian Y, Lu XY. Research progress on recombination based on cloning methods. Acad Period Farm Prod Proc, 2011, 7(6): 7–13 (in Chinese).
戴倩, 田云, 卢向阳. 同源重组介导的克隆策略研究进展. 农产品加工, 2011, 7(6): 7–13.
[19] Chen R, Hu Z, Zhang H. A novel method for constructing small RNA expression vector—artificial pre-mircoRNA. Biotech Bull, 2010, 26(7): 101–105 (in Chinese).陈锐, 胡正, 张辉. 小分子RNA表达载体构建的新方法—MicroRNA前体PCR置换法. 生物技术通报, 2010, 26(7): 101–105.
[20] Zhang XH, Sun YR. The research of homologous recombination in plants. Chin Bull Bot, 2000, 17(2): 137–140 (in Chinese).
张秀海, 孙勇如. 植物基因同源重组研究现状. 植物学通报, 2000, 17(2): 137–140.
[21] Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res, 2008, 18(1): 99–113.
[22] ChenXJ. Mechanism of homologous recombination and implications for aging-related deletions in mitochondrial. Microbiol Mol Biol Rev, 2013, 77(3): 476–496.
[23] Dijon M, Celer CT, Moreau T. Expression and recombination of the EGFP and EYFP genes in lentiviral vectors carrying two heterologous promoters. Cytotherapy, 2005, 7(5): 417–426.
[24] Liddell LC, Manthey GM, Owens SN. Alleles of the homologous recombination gene, RAD59, identify multiple responses to disrupted DNA replication in saccharomyces cerevisiae. BMC Microbiol, 2013, 13: 229.
[25] Vasquez KM, Marburger K, Intody Z, et al. Manipulating the mammalian genome by homologous recombination. Proc Natl Acad Sci USA, 2001, 98(15): 8403–8410.
[26] Mcfadden J. Recombinationin mycobacterial. Mol Microbiol, 1996, 21(2): 205–211.
[27] Li M, Yang Q. A rapid method for generation of homologous recombinant fragments—Fusion PCR, China Biotechnol, 2007, 27(8): 53–58 (in Chinese).李敏, 杨谦. 一种高效构建同源重组DNA片段的方法—融合PCR. 中国生物工程杂志, 2007, 27(8): 53–58.
[28] Szewczyk E, Nayak T, Oakley CE. Fusion PCR and gene targeting in. Nat Protoc, 2006, 1(6): 11–20.
[29] Cha-Aim K, Hoshida H, Fukunaga T. Fusion PCR via novel overlap sequences. Methods Mol Biol, 2012, 852: 97–110.
[30] Ouyang P, Li Y, Yan H. A novel method of constructing vectors: one-step cloning. J Hubei Univ: Nat Sci, 2007, 29(2): 178–181 (in Chinese).欧阳平, 李玉, 严红. 一种载体构建的新方法:一步克隆法. 湖北大学学报: 自然科学版, 2007, 29(2): 178–181.
[31] Kuang FT, Yuan DY, Li L. A novel method for vector making: recombination-Fusion PCR. Genomics Appl Biol, 2012, 31(6): 634–639 (in Chinese). 邝翡婷, 袁定阳,李莉. 一种载体构建的新方法: 重组融合PCR法. 基因组学与应用生物学, 2012, 31(6): 634–639.
[32] Zhang BZ, Ran DL, Liu DB. One-step site-directed mutagenesis based on homologous recombination and dream design. China Biotechnol, 2008, 28(11): 77–81 (in Chinese).张宝中, 冉多良, 刘大斌. 利用DREAM设计和同源重组进行一步定点突变. 中国生物工程杂志, 2007, 28(11): 77–81.
[33] Zhu BG, Cai GF, Emily O. In-FusionTMassembly: seamless engineering of multidomain fusion proteins, modular vectors, and mutations. Biotechniques, 2007, 43(3): 354–359.
[34] Berrow NS, Alderton D, Sainsbury S, Et Al. A versatile ligation-independent cloning method suitable for high-throughput expression screening applications. Nucleic Acids Res, 2007, 35(6): E45.
[35] Berrow NS, Alderton D, Owens RJ. The precise engineering of expression vectors using high-throughput In-Fusion PCR cloning. Methods Mol Biol, 2009, 498: 75–90.
[36] Sean SC, Bartley BA, Lieviant JA. In-Fusion biobrick assembly and re-engineering. Nucleic Acids Res, 2010, 38(8): 2624–2636.
[37] Zhang MJ, Yang J, Zhu CZ. Expression and purification of truncated fragment of extracellular segment of sodium pump α3 subunit in escherichia coli by In-Fusion technology. J Sichuan Univ: Med Sci Edi, 2009, 40(2): 203–207 (in Chinese).张明娟, 杨军, 朱参战. In-Fusion技术构建表达、纯化钠泵Α3亚单位膜外区截断性片段重组质粒. 四川大学学报: 医学版, 2009, 40(2): 203–207.
[38] Liu C, Tang B, Zeng Y. Construction of co-expression vectors of dsred and wild type NAV1.1 or mutated NAV1.1 using In-Fusion technology. J Prac Med, 2011, 27(11): 1908–1910 (in Chinese).刘超, 汤斌, 曾杨. In-Fusion技术构建Ⅰ型钠通道与红色荧光蛋白共表达载体及其突变载体. 实用医学杂志, 2011, 27(11): 1908–1910.
[39] Lin CG, Fan L, Chen LL. Value of In-Fusion cloning techhique on routine vector construction. Chin J Cardiovasc Rehab Med, 2010, 19(1): 10–14 (in Chinese).林朝贵, 范林, 陈良龙. 利用In-Fusion技术构建存活素-增强型绿色荧光蛋白融合基因重组慢病毒表达载体. 心血管康复医学杂志, 2010, 19(1): 10–14.
[40] Han K, Weng JF, Hao ZF. A rapid and high efficient method to construct plant expression vectors. J Maize Sci, 2012, 20(1): 61–66 (in Chinese). 韩凯, 翁建峰, 郝转芳. 一种快速高效构建植物表达载体的方法. 玉米科学, 2012, 20(1): 61–66.
[41] Heckman KL, Pease LR. Gene splicing and mutagenesis by PCR driven overlap extension. Nat Protoc, 2007, 2(4): 924–932.
[42] Li MZ, Elledge SJ. Harnessing homologous recombinationto generate recombinant DNA via SLIC. Nat Methods, 2007, 4(3): 251–256.
[43] Wang ZS, Xiang QJ, Wang HY. Cloning and optimizing expression of a periplasmic solute-binding gene gsiB from. Hereditas (Beijing), 2010, 32(5): 505–511 (in Chinese). 王中山, 向泉桔, 王海燕, 等. 大肠杆菌细胞周质底物结合蛋白Gsib基因的克隆及其表达条件的优化. 遗传, 2010, 32(5): 505–511.
[44] Mamie ZL, Stephen JE. Harnessing homologous recombinationto generate recombinant dna via SLIC. Nat Methods, 2007, 3(4): 251–256.
[45] Bai F, Liu X, Zhang YZ. Liability of a novel gene cloning method: sequence and ligation independent cloning. J Sichuan Univ: Nat Sci, 2008, 45(A Supplementary issue): 33–36 (in Chinese).白帆, 刘珣, 张义正. 一种新的基因克隆方法SLIC的可靠性分析. 四川大学学报: 自然科学版, 2008, 45(增刊): 33–36.
[46] Wang GJ, He C, Zhang YZ. Improvement of the sequence and ligation-independent cloning (SLIC) method. J Sichuan Univ: Nat Sci Ed, 2010, 47(4): 893–898 (in Chinese).王广珺, 何川, 张义正. 不侬赖基因序列和连接反应克隆(SLIC)方法的改进. 四川大学学报:自然科学版, 2010, 47(4): 893–898.
[47] Jeong JY, Yim HS, Ryu JY, et al. One step sequenceand ligationindependent cloning (SLIC): rapid and versatile cloning method for functional genomics studies. Appl Environ Microbiol, 2012, 78(15): 5440–5443.
[48] Gibson DG, Young L, Chuang RY, et al. Enzymatic Assembly of DNA molecules up to several hundred kilobases. Nat Methods, 2009, 6(5), 343–345.
[49] Ellis T, Adie T, Baldwin GS. DNA assembly for synthetic biology: from parts to pathways and beyond. Integr Biol, 2011, 3(2): 109–118.
[50] Sleight SC, Sauro HM. Randomized BioBrick Assembly: a novel DNA assembly method for randomizing and optimizing genetic circuits and metabolic pathways. Acs Synth Biol, 2013, 2(9): 506–518.
[51] Bartosiak-Jentys J, Hussein AH, Lewis CJ, et al. Modular system for assessment of glycosylhydrolase secretion in geobacillus thermoglucosidasius. Microbiol, 2013, 159: 1267–1275.
[52] Wagner JC, Goldfless SJ. Ganesan SM, et al. An integrated strategy for efficient vector construction and multi-gene expression in plasmodium falciparum. Malaria J, 2013, 12(1): 373–390.
[53] Gibson DG, Smith HO, Hutchison CA, et al. Merryman. Chemical synthesis of the mouse mitochondrial genome. Nat Methods, 2010, 7(11): 901–903.
[54] Stefan DK, Leslie HS, Todd S, et al. Rapid and reliable DNA assembly via ligase cycling reaction. ACS Synth Biol, 2014, 3(2): 97–106.
[55] Binder A, Lambert J, Morbitzer R, et al. A modular plasmid assembly kit for multigene expression, gene silencing and silencing rescue in plants. PLoS ONE, 2014, 9(2): E88218.
[56] Engler C, Kandzia R, Marillonnet S. A one pot, one step, precision cloning method with high throughput capability.PLoS ONE, 2008, 3(11): E3647.
[57] Engler C, Gruetzner R, Kandzia R, et al. Golden Gate shuffling: a one-pot DNA shuffling method based on type IIS restriction enzymes. PLoS ONE, 2009, 4(5): E5553
[58] Engler C, Marillonnet S. Generation of families of construct variants using Golden Gate shuffling. Methods Mol Biol, 2011, 729: 167–181.
[59] Morbitzer R, Elsaesser J, Hausner J, et al. Assembly of customtale-type dna binding domains by modular cloning. Nucleic Acids Res, 2011, 39(13): 5790–5799.
[60] Hillson NJ, Rosengarten RD, Keasling JD. j5 DNA assembly design automation software. ACS Synth Biol, 2012, 1(1): 14–21.
[61] Binder A. Lambert J. Morbitzer, R, et al. A modular plasmid assembly kit for multigene expression, gene silencing and silencing rescue in plants. PLoS ONE, 2014, 9(2): E88218
[62] Weber E, Engler C, Gruetzner R, et al. A modular cloning system for standardized assembly of multigene constructs. PLoS ONE, 2011, 6(2): E16765.
[63] Werner S, Engler C, Weber E, et al. Fast track assembly of multigene constructs using Golden Gate cloning and the MoClo system. Bioeng Bugs, 2012, 3(1): 38–43.
[64] Sarrion-Perdigones A, Falconi EE, Zandalinas SI, et al. Goldenbraid: an iterative cloning system for standardized assembly of reusable genetic modules. PLoS ONE, 2011, 6(7): E21622.
(本文责编 陈宏宇)
Methods for construction of transgenic plant expression vector: a review
Yangpu Zhang, and Shushen Yang
,,712100,,
Construction of recombinant plasmid vector for gene expression is a key step in making transgenic plants and important to study gene function and plant genetic engineering. A right choice of gene construction method can be cost-effective and achieve more diverse recombinant plasmids. In addition to the traditional methods in construction of plant gene expression vectors, such as Gateway technology, three DNA method and one step cloning, a few novel methods have been developed in recent years. These methods include oligonucleotide synthesis-based construction of small fragment gene expression vectors via competitive connection; construction of small RNA expression vector using pre-microRNA; recombination-fusion PCR method which inserts DNA fragments of multiple restriction sites into the target vector; and insertion of a DNA fragment into any region of a linear vector via In-Fusion Kit. Construction of complex vectors with many fragments uses sequence and ligation-independent cloning method, Gibson isothermal assembly or Golden Gate assembly. This paper summarizes our working experience in the area of recombinant vector construction and reports from others with an intention to disseminate ideas about currently widely used DNA recombination methods for plant transformation.
artificial pre-Mirco RNA, in-fusion kit, recombination-fusion PCR, Gibson isothermal assembly, Golden Gate assembly
May 6, 2014; Accepted: August 8, 2014
Shushen Yang. Tel: +86-29-87092262; E-mail:yangshushen2002@163.com
Supported by:National Natural Science Foundation of China (No. 31271625), State Key Laboratory of Soil Erosion and Dry land Farming on the Loess Plateau Foundation (No. 10502).
国家自然科学基金(No. 31271625),黄土高原土壤侵蚀与旱地农业国家重点实验室专项(No. 10502) 资助。
猜你喜欢
环球时报(2022-09-20)2022-09-20
华人时刊(2022年9期)2022-09-06
汉字汉语研究(2021年2期)2021-08-30
今日农业(2020年24期)2020-12-15
华人时刊(2020年15期)2020-12-14
汉字汉语研究(2019年2期)2019-08-27
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
河北书画研究(2016年3期)2016-04-28
广州大学学报(自然科学版)(2015年4期)2015-12-23
小资CHIC!ELEGANCE(2015年15期)2015-09-01
