
10例Gitelman综合征患者SLC12A3基因的检测
2015-06-09 06:49韩正斌邵乐平
肾脏病与透析肾移植杂志 2015年5期
王 艳 韩正斌 邵乐平 栾 健 刘 军
10例Gitelman综合征患者SLC12A3基因的检测
王 艳1韩正斌1邵乐平1栾 健2刘 军3
目的:对青岛地区10例可疑Gitelman综合征患者致病基因SLC12A3和CLCNKB的突变特点进行分析。 方法:通过直接测序的方法进行突变分析。选取100例健康人作为对照。 结果:共确定SLC12A3基因9个突变位点,其中3个为新突变位点,包括2个错义突变:Glu429Lys,Ala264Gly;1个缺失突变:1740delC。6个已报道过的突变,其中包括5个错义突变:Cys430Gly,Asp486Asn,Ser283Tyr,Thr163Met,Arg913Gln;1个缺失突变:2877_2878delAC。10例患者中8例携带Ala264Gly纯合突变,4例携带Asp486Asn杂合突变,大部分患者为复合杂合突变。 结论:本研究共发现9个突变位点,其中3个新突变位点。
Gitelman综合征 突变 SLC12A3基因
Gitelman综合征(GS)是一种常染色体隐性遗传性疾病(OMIN 263800),为肾单位远曲小管重吸收Na-Cl障碍造成的原发肾性失盐性疾病。GS是由SLC12A3 基因失活突变导致,该基因其编码噻嗪类利尿剂敏感的Na-Cl共同转运子(NCCT)。SLC12A3基因(MIM 600968)位于染色体16q13,大小约55 kb,包括26个外显子。NCCT由1 021个或1 030个氨基酸残基组成,包括12个跨膜部分(S),一个较大的细胞内N端和一个疏水的C端[1]。以往GS被认为是Bartter综合征(BS)的一个亚型,且经典型BS同GS表型存在重叠和交叉,故临床上很容易混淆。同时,研究表明,少数疑似为GS的患者是由CLCNKB基因突变引起[2],存在遗传异质性,因此本研究分析对10例GS 患者的SLC12A3和CLCNKB基因,以从分子水平提高对该病的认识。
对象和方法
研究对象 本研究组选取2010年7月至2013年7月青岛大学医学院附属医院肾内科住院治疗的10例疑似GS患者,其中男性7例,女性3例,平均年龄(32±13)岁,分别来自9个不同的汉族家系。诊断标准:慢性持续性低血钾(<3.5 mmol/L)、低血镁(<0.65 mmol/L)和低尿钙(24h尿钙/尿肌酐<0.1 mmol/mmol)。所有患者均无长期服用缓泻剂、利尿剂及药物成瘾史。选取100例非相关的健康者作为对照组,评估本研究发现的新突变位点。本研究获得青岛大学医院附属医院伦理委员会的批准,所有参与者均签署知情同意书。
研究方法
外周血基因组DNA提取 采用试剂盒(GenElute,Sigma NA2010)抽取患者及其他家庭成员外周血基因组DNA。
引物设计 SLCl2A3基因序列来自Genbank(NC_000016),采用Primier 5软件自行设计23对引物扩增SLCl2A3基因全部26个外显子及其侧翼内含子序列[3]。引物由上海生物工程技术服务有限公司合成。根据Simon等[4]的研究设计19对引物扩增CLCNKB基因全部19个外显子。
PCR产物扩增 在25 μl的反应体系中包含0.2 mmol/L dNTP、0.03 U/ml Taq 聚合酶(DRR006B,大连宝生)、2.0 mmol/L MgCl2、2.5 ml 10X PCR Mg2+free Buffer(Takara)、30 ng基因组DNA以及各1 mmol/L上下游引物。在梯度PCR仪上95℃变性45s,59℃~71℃退火45s,72℃延伸45s,33个循环后72℃最后延伸10 min。
PCR产物纯化和测序 采用磷酸虾碱酶和外切酶Ⅰ纯化PCR产物,ABI Prism 3700 DNA分析仪(美国Applied Biosystems)进行单向或双向测序。
DNA测序结果分析 采用Vector NTI11.0与正常序列比对。
为预测可能发现的错义突变的致病性 我们引入了错义突变致病性的评分系统[5-6],并采用Vector NTI advance 10-Align对8种SLC12A3和CLCNKB基因编码的同源蛋白进行序列比对。
结 果
临床表现和实验室检查结果 10例患者中,9例表现骨骼肌功能障碍(肌肉无力、麻木、搐搦、瘫痪),1例无症状,在查体中发现低血钾。5例患者血浆醛固酮水平升高,7例血浆肾素活性升高,10例有低尿钙(24h尿钙/尿肌酐<0.1 mmol/mmol)(表1)。
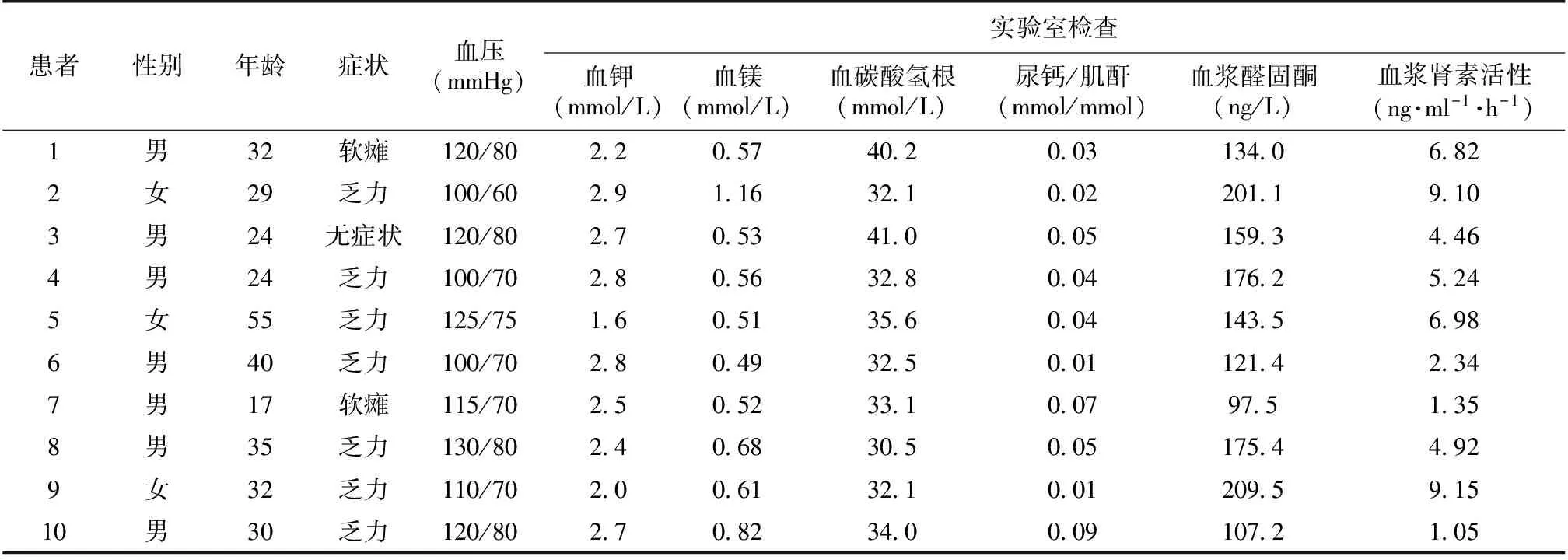
表1 10例患者的临床表现和实验室检查
基因分析 通过直接测序,10例患者中共发现SLCl2A3基因的9个突变位点可能与GS相关,其中包括7个错义突变,2个移码突变。在对照组中未发现此9个突变。全部突变位点中,6个突变位点(Cys430Gly, Asp486Asn,Ser283Tyr,Thr163Met,Arg913Gln, 2877_2878delAC)已有报道[6-7];经检索人类基因突变库(HGMD)和最近文献,3个突变位点被证实是本研究发现的新突变,其中包括2个错义突变:Glu429Lys,Ala264Gly;1个缺失突变:1740delC(图1)。所有突变点在NCCT中的位置见图2。所有患者均未发现CLCNKB基因的突变。
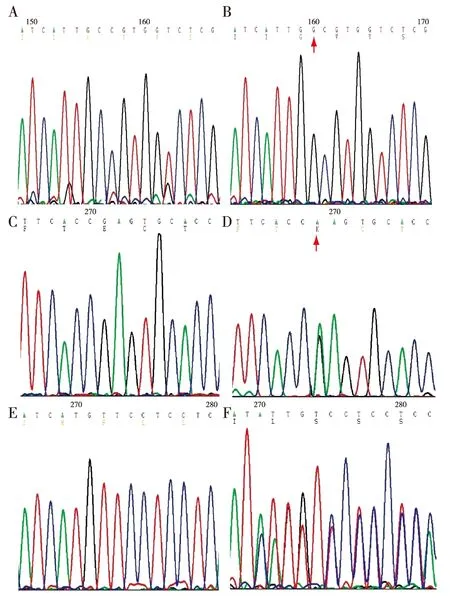
图1 新发现突变位点基因图谱A:Ala264Gly的野生型;B:Ala264Gly的突变型;C:Glu429Lys的野生型;D:Glu429Lys的突变型;E:1740delC的野生型;F:1740delC的突变型;Ala264Gly中碱基G突变为A,编码的氨基酸相应的由Ala变成Gly;Glu429Lys中碱基G突变为A,编码的氨基酸相应的由Glu变成Lys;而1740delC在1740为缺失碱基C,引起框移突变
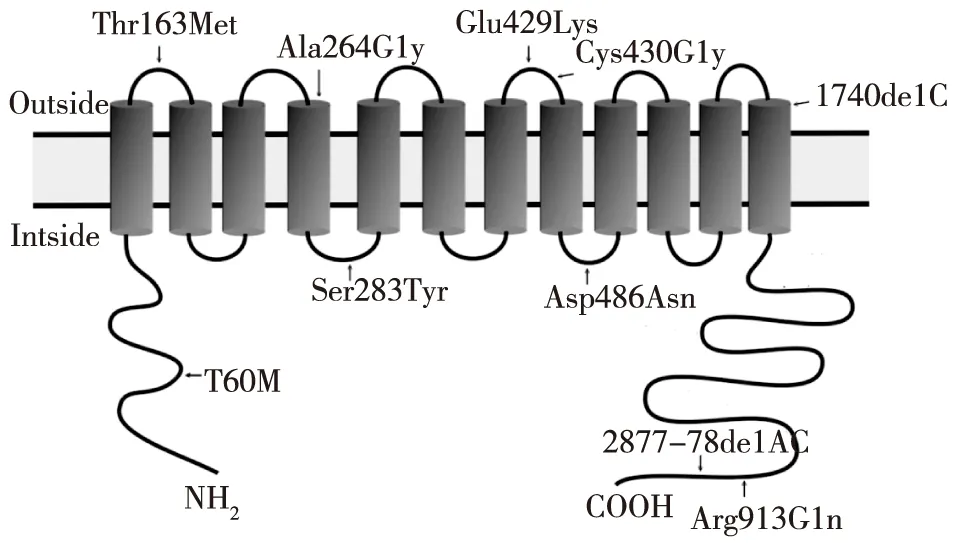
图2 突变点在Na-Cl共同转运子中的位置示意图
除1例患者仅发现1个突变位点外,其余9例患者均存在≥2个突变点,包括纯合突变3个,杂合突变6个,携带纯合突变患者的父母无近亲婚配,所有携带杂合型突变患者非近亲婚配子女。8例患者发现纯合突变Ala264Gly。4例患者携带Asp486Asn杂合突变。家系A中,患者6携带有杂合突变Glu429Lys和1740delC及纯合突变Ala264Gly,但其子例7仅遗传了父亲的杂合突变1740delC和纯合突变Ala264Gly,未遗传杂合突变Glu429Lys。1740delC提前终止于第610位密码子。例5发现的2877_2878delAC突变位点已有报道[7-8],但其报道的为杂合突变,本研究发现的为纯合突变,较为少见(表2)。
对本研究发现的7个错义突变位点的致病性分析,P.C430G、p.S283Y和p.T163M存在高致病可能,p.264G和p.D486N中度致病可能,p.E429K和p.R913Q可能不致病(表3)。同源序列比较(Homosapiens (NP_000330.2)、Bos taurus(NP_0 ̄0 ̄1 ̄1 ̄9 ̄3 ̄1 ̄0 ̄7.1)、Canis lupus familiaris(XP_535292.3)、Danio rerio(NP_001038545.1)、Gallus gallus(XP_0 ̄0 ̄4 ̄9 ̄4 ̄4 ̄1 ̄8 ̄9.1)、Mus musculus(NP_001192240.1)、Rattus norvegicus(NP_062218.3),和Xenopus(Silurana)tropicalis(XP_004913583.1)),显示NCCT第163、264、430、486位氨基酸在8种生物种均高度保守,283和429分别在6和5种生物中保守。而913位的Arg在其他3种生物中是Gln(图4)。
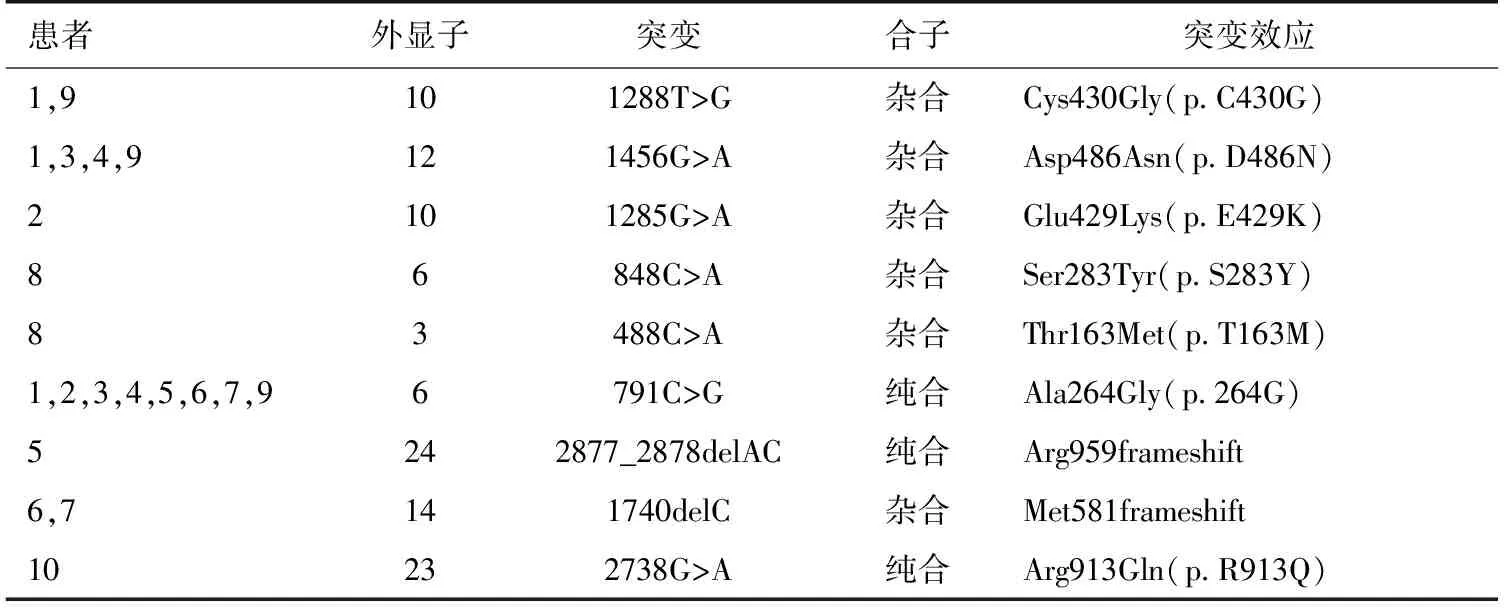
表2 10例患者的突变位点
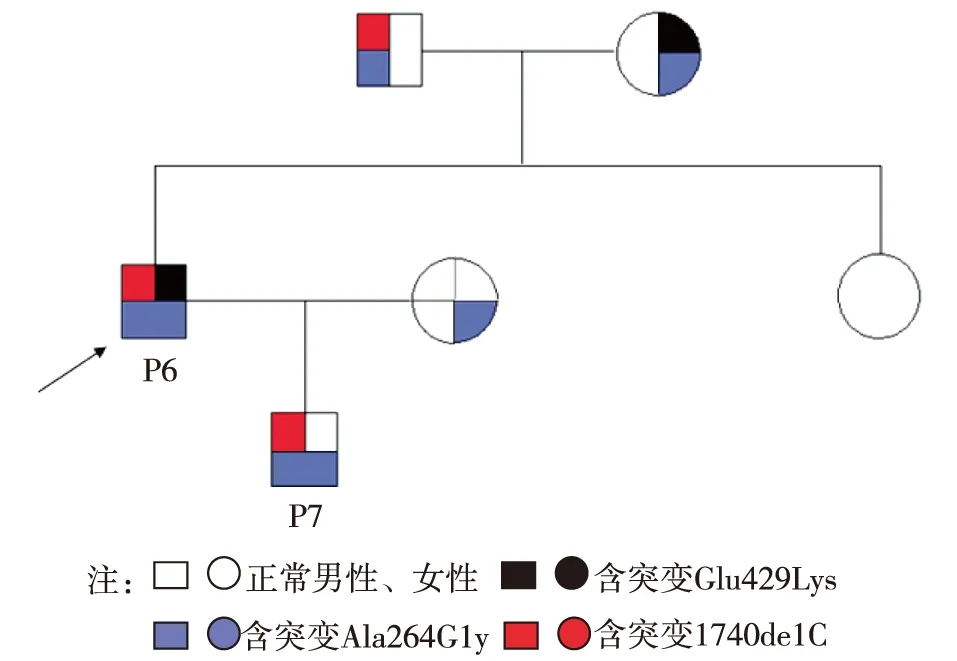
图3 家系A遗传图谱
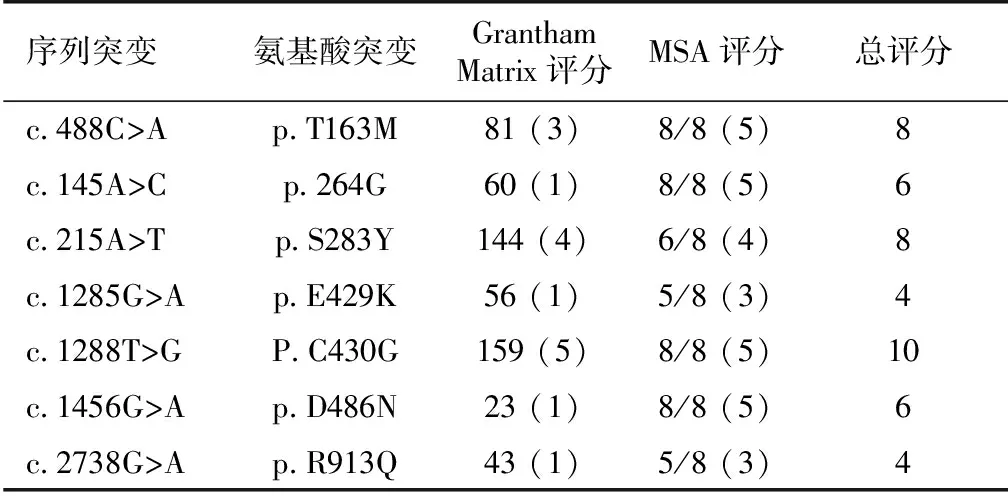
表3 错义突变致病性评分
Grantham Matrix评分(括号内):<60:1分; 61~78.3:2分; 78.4~93.4:3分;>93.4:4分;任何半胱氨酸的替代=5分;多重序列比对评分(MSA评分)(括号内):8种生物全部保守=5;6~7=4;4~5=3;1~3=1;总分≥8:高致病可能;6~8:中度致病可能;≤5:可能非致病(例如多态性)
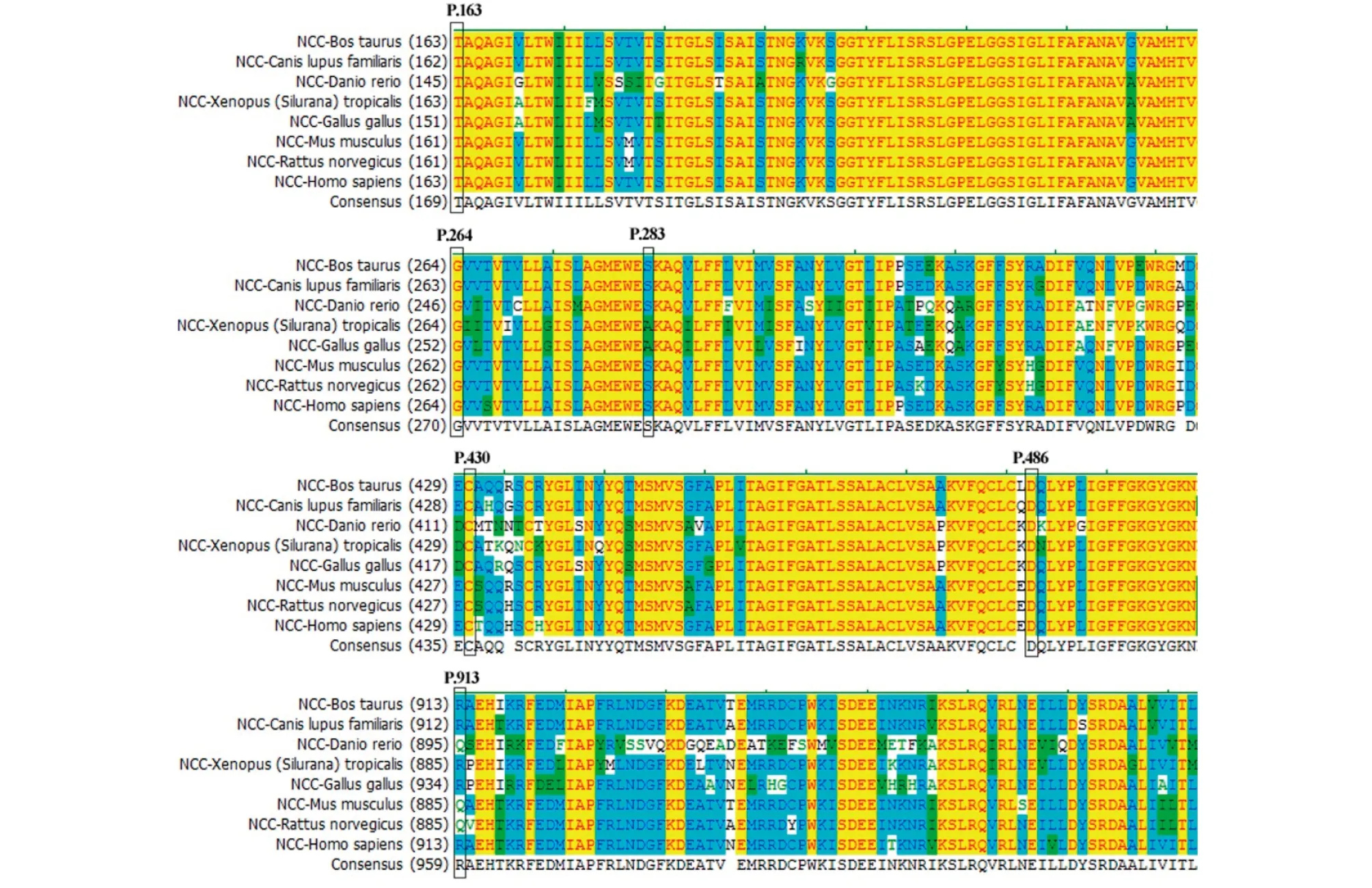
图4 错义突变多重序列比对评分(MSA)图
讨 论
GS以低血钾、低血镁、低尿钙、代谢性碱中毒、高肾素-血管紧张素-醛固酮及正常或偏低的血压为特点。其中低尿钙、低血镁在临床上被认为是GS和BS的主要区别点,但这并非完全可靠[9]。随着分子遗传学的发展,突变基因的发现,给GS的确诊提供了更为可靠的证据。自1996年Simon等[4]从分子角度第一次确认GS的突变基因(SLC12A3)不同于BS(CLCNKB)以来,迄今为止,已经发现有100多个突变基因与GS有关[10-11]。通过GS致病基因的研究,不仅可以加深对该病的临床认识及病理生理和分子发病机制的理解,同时也可以为该病的诊断提供了更为可靠的依据。因此,本研究通过分析10例GS综合征患者的基因突变情况,以期进一步提高对该病的认识和丰富SLC12A3突变基因库。
本研究通过直接测序,在10例患者中共发现SLC12A3基因9个突变位点可能与GS有关,其中3个位于NCCT胞外段(163、429、430),4个位于胞内段(283、486、913,2877),2个位于跨膜区(264,1740)。本研究未发现突变位点在结构域分布上与疾病之间的关系,与国外文献一致[12]。在此次发现的错义突变中,MSA图示NCCT第163、264、430、486位氨基酸在8种生物种均高度保守,提示Thr163Met、Ala264Gly、Cys430Gly、Asp486Asn可能均是很有意义的突变;NCCT第283位氨基酸虽然仅在6种物种上同源,但Ser283Tyr错义突变致病性总评分8分,仍提示高度致病性;对于Arg913Gln和Glu429Lys,虽错义突变致病性总评分均为4分,且第913位的Arg在其他3种生物中是Gln,因此Arg913Gln和Glu429Lys很可能是多态性突变。而1740delC和2877_2878delAC均引起框移突变,产生截短的或异常的翻译产物,对NCCT的结构和功能影响较大,很可能是很有意义的致病性突变。然而,值得指出的是,采用爪蛙卵母细胞、cos-7细胞等生物体,选取以上突变位点,采用蛋白标签、定点突变等技术,进行功能表达研究较错义突变致病性的评分系统及基因编码的同源蛋白进行序列比对更为准确[12]。
本研究大多数患者就诊主诉为骨骼肌功能障碍症状,如乏力、抽搐甚至瘫痪。但例3却无明显症状,仅在查体中发现低钾。这提示我们,并非所有GS患者均有临床症状。有文献曾报道一个家系多人携带SLC12A3基因中的相同突变,但临床表现迥异[13-14]。国内学者亦曾对GS的高频突变基因型和表型的相关性进行研究,结果为无明显相关性[14]。因此,典型GS表型的出现,可能是多个突变基因协同作用的结果;同时,后天的环境、饮食的影响亦不能忽视。10例GS患者,女性仅3例,远少于男性。欧洲和台湾学者均发现[15-16],男性GS患者较女性症状更重,就诊率高;同时,动物实验也证实[17],正常雌性SD大鼠肾脏表达的NCCT受体的密度是雄性大鼠的2倍,切除卵巢能显著减少NCCT受体的密度。以上证据均提示性别是导致表型多样性的因素之一。
有研究显示,Thr60Met在正常亚洲人群中发现该突变位点的突变频率为0.5%~0.8%[15],而在GS患者中的突变频率高达33%,推测Thr60Met可能是亚洲人群特有的高发突变。但本研究却未发现该突变点,而突变点Ala264Gly的频率却高达38%(8/21),考虑可能原因为本研究所涉及样本量较少,与相对较大的样本比较,结果存在偏差。
既往文献报道,高达40%左右的GS患者仅发现1个突变位点[16],可能原因有:(1)突变位点可能位于SLCl2 A3基因的调节序列或内含子的深部,而上述两部分平时不予测序分析;(2)包含1个或多个外显子的基因片断重排后难以通过单个外显子分析的方法确定;(3)突变可能位于其他调节NCCT功能的基因,如WNKl,WNK4等[15]。虽然未针对上述可能原因在基因测序方面做相应改进,但本研究中仅10%(1/10)的患者存在一个突变位点,远远低于文献报道,考虑可能原因为本研究样本量较 2个突 S为一种常染色体隐性遗传病,根据遗传定律,除非是近亲结婚或者婚配同样的患者,一个家系连续两代均出现患者极为罕见[18]。但本研究中,家系A中父子2人均为GS患者。追问婚姻史,例7父母并非近亲结婚。由此推测,例6的配偶有可能也是GS患者,因无症状而未就诊;而例7患者携带的纯合突变Ala264Gly其中之一即来自其母亲。因此,这似乎从一定程度上说明,GS并非罕见病,其发病率可能远远大于既往文献报道。同时,Ala264Gly可能确为本区域GS的热点突变。
综上所述,通过直接测序,我们共发现GS致病基因SLCl2A3的9个突变位点,其中3个为新突变位点;典型GS表型的出现,可能是多个突变基因协同作用的结果;通过GS致病基因的研究,不仅可以加深对该病的分子发病机制的理解,同时也可以为该病的诊断提供更为可靠的依据。
1 Galli-Tsinopoulou A,Patseadou M,Hatzidimitriou A,et al.Gitelman syndrome:first report of genetically established diagnosis in Greece.Hippokratia,2010,14(1):42-44.
2 Zelikovic I,Szargel R,Hawash A,et al.A novel mutation in the chloride channel gene, CLCNKB, as a cause of Gitelman and Bartter syndromes.Kidney Int,2003,63(1):24-32.
3 邵乐平,任红,王伟铭,等.Gitelman综合征SLCl2A3基因突变研究.中华肾脏病杂志,2007,23(6):351-356.
4 Simon DB,Bindra RS,Mansfield TA,et al.Mutations in the chloride channel gene,CLCNKB,cause Bartter’s syndrome type Ⅲ.Nat Genet,1997,17(2):171-178.
5 Grantham R.Amino acid difference formula to help explain protein evolution. Science,1974,185(4154):862-864.
6 Abkevich V,Zharkikh A,Deffenbaugh AM, et al.Analysis of missense variation in human BRCA1 in the context of interspecific sequence variation.J Med Genet,2004,41(7):492-507.
7 Shao L,Ren H,Wang W,et al.Novel SLC12A3 mutations in Chinese patients with Gitelman syndrome.Nephrol Physiol,2008,108(3):29-36.
8 Hsu YJ,Yang SS,Chu NF,et al.Heterozygous mutations of the sodium chloride cotransporter in Chinese children:prevalence and association with blood pressure.Nephrol Dial Transplant,2009,24(4):1170-1175.
9 Knoers NV,Levtchenko EN.Gitelman syndrome.Orphanet J Rare Dis,2008,3:22.
10 Hsu YJ,Yang SS,Chu NF,et al.Heterozygous mutations of the sodium chloride cotransporter in Chinese children:prevalence and association with blood pressure.Nephrol Dial Transplant,2009,24(4):1170-1175.
11 Reissinger A,Ludwig M,Utsch B,et al.Novel NCCT gene mutations as a cause of Gitelman syndrome and a systematic review of mutant and polymorphic NCCT alleles.Kidney Blood Press Res,2002,25(6):354-362.
12 Miao Z,Gao Y,Bindels RJ,et al.Coexistence of normotensive primary aldosteronism in two patients with Gitelman′s syndrome and novel thiazide-sensitive Na-Cl cotransporter mutations.Eur J Endocrinol,2009,161(2):275-283.
13 Ueda K,Makita N,Kawarazaki H,et al.A novel compound heterozygous mutation of Gitelman′s syndrome in Japan,as diagnosed by an extraordinary response of the fractional excretion rate of chloride in the trichlormethiazide loading test.Intern Med,2012,51(12):1549-1553.
14 秦岭,邵乐平,任红,等.中国人Gitelman综合征高发突变的基因型和表型特征.肾脏病与透析肾移植杂志,2008,17(8):331-334.
15 Lin SH,Shiang JC,Huang CC,et al.Phenotype and genotype analysis in Chinese patients with Gitelman′s syndrome.J Clin Endocrinol Metab,2005,90(5):2500-2507.
16 Riveira-Munoz E,Chang Q,Bindels RJ,et al.Gitelman syndrome:towards genotype-phenotype correlations? Pediatr Nephrol,2007,22(3):326-332.
17 Chen Z,Vaughn DA,Fanestil DD.Influence of gender on renal thiazide diuretic receptor density and response.J Am Sec Nephrol,1994,5(4):1112-1119.
18 Yagi H,Yahata K,Usui T,et al.Inheritance of an autosomal recessive disorder,Gitelman′s syndrome,across two generations in one family.Intern Med,2011,50(11):1211-1214.
(本文编辑 春 江)
Analysis of SlCl2A3 gene mutation in patients with Gitelman syndrome
WANGYan1,HANZhengbin1,SHAOLeping2,LUANJian1,LIUJun3
DepartmentofNephrology,theAffiliatedHospitalofMedicalCollege,QingdaoUniversity,Qingdao266003,China2DepartmentofEndocrinology,theQingdaoMunicipalHospital(Group),Qingdao266003,China3DepartmentofNephrology,theQingdaoNO.8people’shospital,Qingdao266003,China
Objective:To identify the new mutations of SLCl2A3 and CLCNKB gene in patients with suspicious Gitelman syndrome, and Analyze the characteristics of the genetic mutations. Methodology:Ten patients hospitalized in the affiliated hospital of Qingdao university with the clinical and biochemical features of Gitelman syndrome were analyzed by direct sequencing of SLCl2A3 gene.One hundred unrelated normal subjects were selected to evaluate all the mutations found by this study and make sure that they are new mutations through the Human Gene Mutation date base. Results:Nine mutations were identified in SLCl2A3 gene of 10 patients with Gitelman syndrome. Four were novel variants, including 2 missense mutations:Glu429Lys, Ala264Gly, and one deletions:1740delC. Six were recurrent ones including 5 missense mutations: Cys430Gly, Asp486Asn, Ser283Tyr, Thr163Met, Arg913Gln; and one deletion: 2877_2878delAC. The Homozygous mutation Ala264Gly was found in 8 of 10 patients while the heterozygous mutation Asp486Asn in 4 of 10.The majority of the patients were compound heterozygous. Conclusion:Nine mutations were identified in SLCl2A3 gene of 10 patients with Gitelman syndrome, and 3 were novel variants.
Gitelman syndrome Mutation SLCl2A3 gene
山东省科技厅基金(2011GSF11832)
1青岛大学医学院附属医院肾内科(青岛,266003);2青岛市市立医院(集团)内分泌科;3青岛市第八人民医院肾内科
ⓒ 2015年版权归《肾脏病与透析肾移植杂志》编辑部所有
2015-03-17
猜你喜欢
广西医科大学学报(2022年5期)2022-06-07
昆明医科大学学报(2022年3期)2022-04-19
种子(2021年3期)2021-04-12
中南医学科学杂志(2019年6期)2019-12-05
中成药(2017年12期)2018-01-19
外语教学理论与实践(2016年1期)2016-06-11
湖南畜牧兽医(2016年3期)2016-06-05
中学生理科应试(2016年7期)2016-05-14
浙江医学(2014年17期)2014-04-13
华东理工大学学报(自然科学版)(2014年1期)2014-02-27
