
利用钛白副产物浸出软锰矿生产硫酸锰的研究
2015-05-30 07:43卢伟胜黄炳龙陈南雄黄冠汉罗昌璃
中国锰业 2015年3期
卢伟胜,黄炳龙,陈南雄,黄冠汉,罗昌璃
(1.中信大锰矿业有限责任公司,广西 南宁 530029;2.中信大锰矿业有限责任公司大新锰矿分公司,广西 大新 532315)
0 前言
锰可用于钢铁、锰铁、有色合金、手机电池和其他化工产品的生产。锰盐对于人体健康也很重要,因为他们可以维持健康的神经系统和免疫系统以及调节血糖。作为一种原材料用于生产染料中锰化合物、药物、纺织品以及不同工业的产品,硫酸锰是锰化合物中产量最大的。从经济上考虑,锰矿浸出生产硫酸锰是非常重要的生产方法[1]。对锰需求的不断增长正导致该资源的快速消耗。因此,大量的注意力被放在提高锰矿和矿渣还原浸出上[2]。
当锰以二价形式存在时,由于其可溶性,对应的锰盐通常直接通过酸浸出获得。当锰以四价形式存在时,需要还原剂将其还原成可溶的含Mn2+的化合物[3]。利用硫酸进行锰矿浸出是常用的方法[4]。不同的还原剂被用来与硫酸一起进行浸出研究,例如天然气、草酸、甲醇、碳水化合物、煤、石墨、二氧化硫、氢、谷物秆等[5-10]。Jana 等[11]研究在硫酸盐还原剂(如SO2、FeS2、Na2SO3)和热水存在的情况下,将锰矿焙烧后进行浸出。Jiang等[12-13]研究锰矿在含有过氧化氢的硫酸溶液进行浸出。Ismail等[14]利用锯末或乳糖作为氧化剂在硫酸中进行低品位锰矿浸出研究。他们指出,直接还原浸出(在溶液中还原浸出)相对于其他还原工艺的优势是还原和浸出步骤可在一步完成。该工艺避免高温焙烧,相对清洁和节能,而且产生的硫酸锰溶液可以在同一个系统中净化。
为了使锰还原尽可能经济,廉价的试剂和高效的技术是必不可少的。利用硫酸亚铁进行锰矿处理已经被多次报道[15-17]。在中国,来自钛白副产物的硫化亚铁相对便宜且产量较大。如何利用这个副产物进行锰矿还原浸出已引起较多关注。本文将利用硫酸亚铁作为还原剂,系统地研究软锰矿还原浸出,从而获得生产高纯一水硫酸锰的优化工艺条件。
1 试验部分
1.1 化学反应
Das等[16]研究发现,低品位锰矿中二氧化锰与硫酸亚铁可有3种反应方式。
1)在中性的硫酸亚铁溶液中,
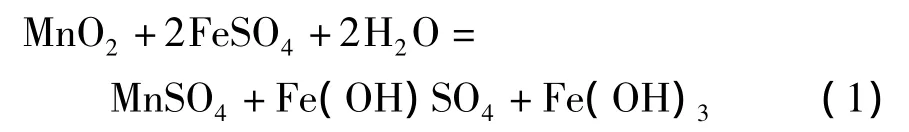
2)在含有少量酸的硫酸亚铁溶液中,
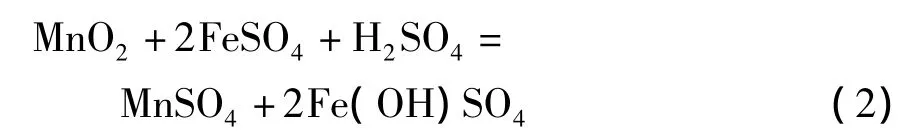
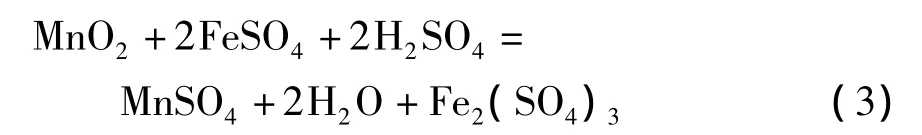
3)在含有过量酸的硫酸亚铁溶液中,多数研究工作集中于方程式(3)的化学反应上。在硫酸亚铁量是理论值的1~1.2倍,反应温度70~95℃,反应时间 2 ~3.5 h,液固比为 3∶1 ~8∶1和初始硫酸浓度为180~210 g/L情况下,软锰矿的锰浸出率可以超过95%。在这种强酸性及还原剂过量的条件下,需要考虑几个问题。首先,需要氧化剂来氧化过量的硫酸亚铁。如果仍然利用软锰矿作为氧化剂,将降低二氧化锰的浸出率。从经济上考虑,也不宜使用双氧水和高锰酸钠作为氧化剂;其次,额外需要碳酸锰或石灰乳来中和浸出液中由于Fe3+水解沉淀产生的酸;在过量酸性或强酸性条件下,硫酸亚铁将迅速被氧化为硫酸铁,使得浸出液中Fe3+浓度非常高。因此,需要加入硫酸钠或硫酸钾使Fe3+大部分以黄钠铁矾[18]或黄钾铁矾形式沉淀,才能使浸出浆液容易过滤,否则形成大量Fe(OH)3胶体,使过滤难以进行。
但是,当浸出浆液含有少量硫酸以及工艺经过精心设计后,可以防止以上问题发生。作为钛白副产物的硫酸亚铁中含有少量硫酸,若按10∶1的液固比将硫酸亚铁溶于水,其溶液的pH值为2.0。因此,本文重点研究利用钛白副产物硫酸亚铁作为还原剂,基于化学方程式(2)的反应过程,以一氧化锰为中和剂控制浸出浆液的pH值在2.0~5.0之间,并控制氧化水解的Fe3+浓度<1 mg/L。采用针铁矿法[19]沉淀Fe3+,不需要添加硫酸钠(或硫酸钾)作为辅助沉铁剂。针铁矿法除铁反应方程式可表述如下:
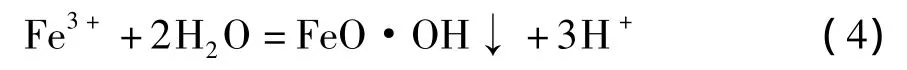
利用一氧化锰将方程式(4)中的酸完全中和,反应方程式如下:
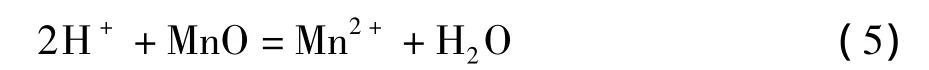
1.2 试验设备和材料
通过能量色散 X射线荧光谱(OXFORD,MDX1000)和扫描电镜(JEOL,FEG-XL 30S)确定软锰矿样品的成分;利用电感耦合等离子体发射光谱(Thermo Scientific,iCAP6300)对所制得的硫酸锰粉末进行杂质分析;经过校准的玻璃电极(Mettler Toledo InLab®420)用于测量浸出浆液的pH值。为了确保测量的精度,对复制的样品进行相同测量。分析误差估计小于5%。
软锰矿的化学成分如表1所示。一氧化锰粉末含34%Mn2+。七水硫酸亚铁(FeSO4·7H2O)—钛白粉厂的副产物,含Fe量为19%。所有试剂都是实验室分析纯级别,去离子水用于溶解和稀释。
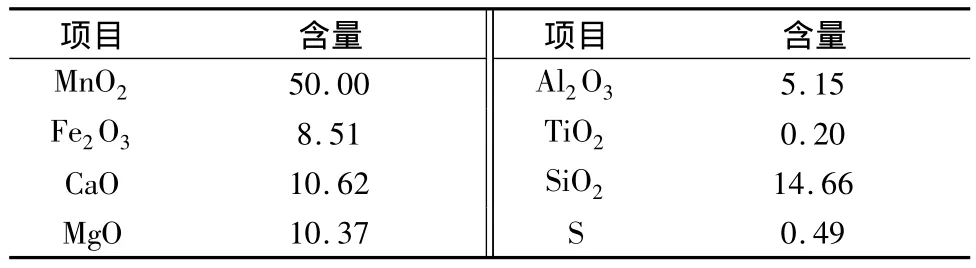
表1 软锰矿化学成分(质量分数)/%
1.3 工艺流程
工艺流程如图1所示。
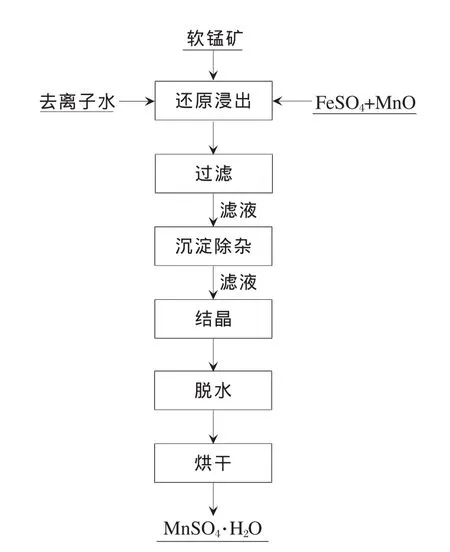
图1 软锰矿浸出生产硫酸锰的工艺流程
将软锰矿样品粉碎,研磨后过筛,按不同粒度分组;在烧杯中,使用去离子水(液固比为5∶1)对样品进行洗涤,以除去任何可溶物质;过滤后,将样品放置在马弗炉内加热至100℃并烘干1 h;浸出试验在密闭的长颈瓶中进行,将七水硫酸亚铁溶解在去离子水中,加热至80℃,然后将100 g处理好的软锰矿样品放入瓶中。一氧化锰粉用于调节浸出料液的pH值;过滤浸出料液,然后对滤液进行沉淀除杂;经过结晶,脱水和烘干,滤液最终成为一水硫酸锰。如果没有特别说明,浸出试验在以下条件中进行:样品粒度范围在 45 ~52 μm,液固比为 3∶1,m(硫酸亚铁)∶m(二氧化锰)∶m(一氧化锰)=2.08∶1∶0.24,反应过程中搅拌浆液速度为400 r/min,反应温度为80℃,反应时间为2 h。
2 试验结果及讨论
2.1 浸出影响因素试验
在本节中,将研究不同因素对浸出过程的影响,这些因素包括软锰矿粒度、搅拌速度、浸出时间、液固质量比、m(硫酸亚铁)∶m(二氧化锰)、pH值和浸出温度。
2.1.1 粒度的影响
将软锰矿样品按粒度分成以下5组,不同粒度下锰回收率的结果见表2。

表2 软锰矿粒度对于锰回收率的影响
由表2可知,随着软锰矿粒度减小,锰回收率增加。但当粒度<45~52 μm时,粒度对锰回收率的增加几乎没有影响,且粒度继续减小,锰回收率下降。基于经济考虑,不需要将软锰矿研磨得过于微细。
2.1.2 搅拌速度的影响
设定软锰矿粒度为45~52 μm,液固质量比为3∶1,m(硫酸亚铁)∶m(二氧化锰)∶m(一氧化锰)=2.08∶1∶0.24,浸出温度为 80℃,浸出时间为 2 h,通过调整搅拌速度在100~600 r/min范围,研究搅拌速度对锰回收率的影响,结果见图2。
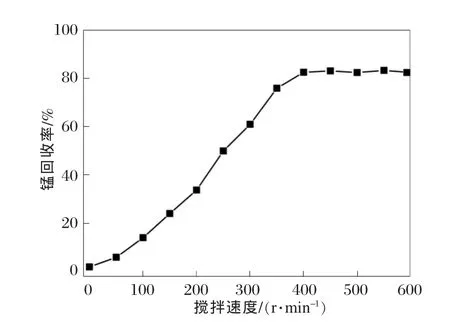
图2 搅拌速度对锰回收率的影响
从图2可以看出:锰回收率随着搅拌速度的增加而增加。这是因为较高的搅拌速度使浆液粘度的影响减小,从而增加反应发生的概率。但是,当搅拌速度超过400 r/min时,锰回收率达到饱和,基本上与搅拌速度无关。这表明在此高速搅拌下,化学反应发生的概率达到最大。在以下的试验中,搅拌速度设为400 r/min。
2.1.3 浸出时间的影响
设定软锰矿粒度为45~52 μm,液固质量比为3∶1,m(硫酸亚铁)∶m(二氧化锰)∶m(一氧化锰)=2.08∶1∶0.24,搅拌速度为 400 r/min,浸出温度为80℃。在20~160 min范围调整浸出时间,研究浸出时间对锰回收率的影响,结果见图3所示。
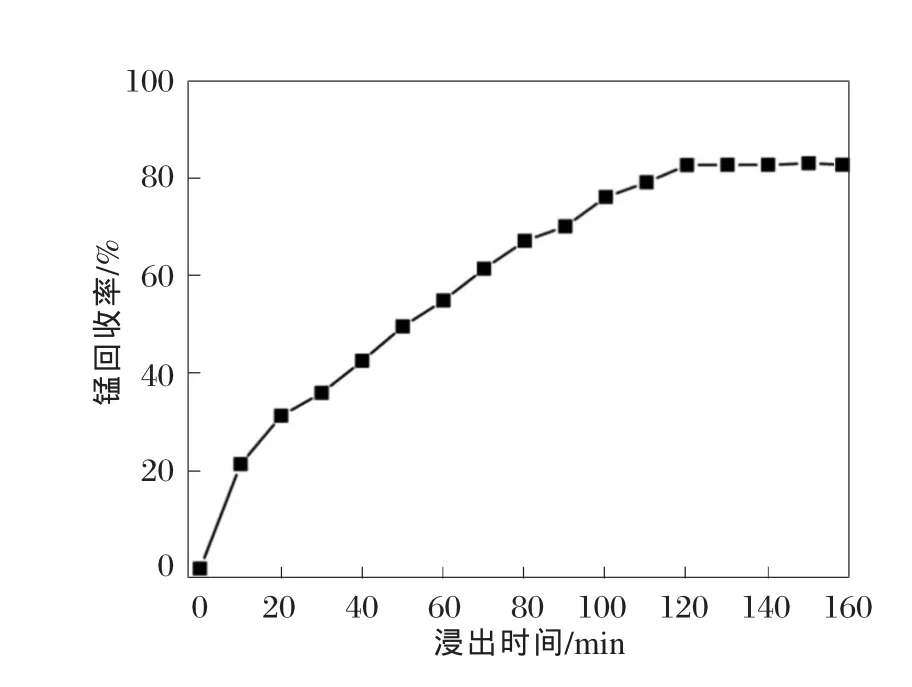
图3 浸出时间对锰回收率的影响
从图3可以看出,当浸出时间超过120 min时,锰回收率几乎不再增加。因此,对于浸出而言,120 min是足够的。值得注意的是,浸出过程在不同时间段以不同的速度在进行,这意味着浸出过程可能涉及多个反应步骤。Ismail等[14]认为通过还原剂对软锰矿进行锰浸出至少涉及4个连续步骤,分别是水解、还原剂从溶液中扩散到固体反应场所(即软锰矿颗粒)、在反应场所形成生成物的晶核、生成物水合作用以及扩散到溶液中。由此看出,浸出过程是与时间有关的过程。
2.1.4 液固质量比的影响
设定软锰矿粒度为45~52 μm,m(硫酸亚铁)∶m(二氧化锰)∶m(一氧化锰)=2.08∶1∶0.24,搅拌速度为400 r/min,浸出温度为80℃,浸出时间为2 h。在1∶1~4∶1范围调整液固质量比,研究液固质量比对锰回收率的影响,结果见图4。
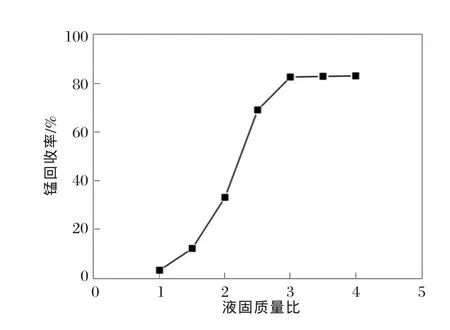
图4 液固质量比对锰回收率的影响
从图4可以看出,在较低的液固质量比范围内,锰回收率较低。这是由于在低液固质量比时,浆液粘度较高,使得软锰矿颗粒粘成一团,导致Fe2+不能达到软锰矿颗粒团的内部进行反应,因此,只有小部分软锰矿颗粒得以参加反应。随着液固质量比的增加,浆液的粘度逐渐降低,使得软锰矿颗粒成团的概率降低,Fe2+可以达到大多数软锰矿颗粒进行反应,所以,更多的锰可以从矿里浸出。当液固质量比>3∶1时,锰回收率几乎没有增加,这意味着大多数锰已经浸出形成硫酸锰溶液[20]。因此,在以下试验中,采用3∶1的液固质量比。
2.1.5 硫酸亚铁与二氧化锰质量比的影响
设定软锰矿粒度为45~52 μm,液固质量比为3∶1,m(二氧化锰)∶m(一氧化锰)=1∶0.24,搅拌速度为400 r/min,浸出温度为80℃,浸出时间为2 h。通过调整硫酸亚铁与二氧化锰质量比,研究其对锰浸出的影响,结果见图5。
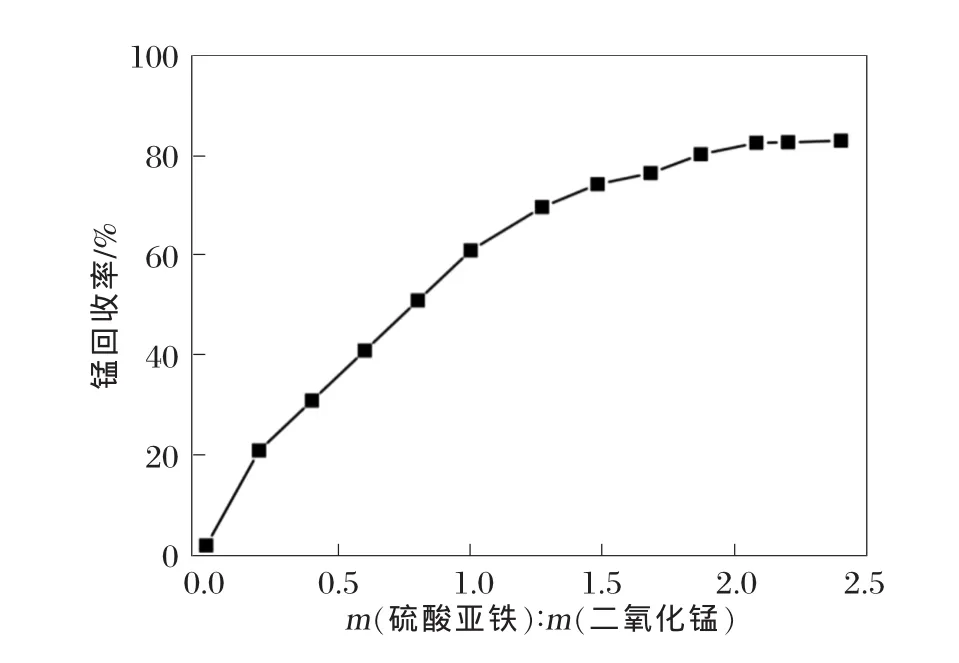
图5 硫酸亚铁与二氧化锰质量比对锰回收率的影响
从图5看出:在锰浸出过程中,硫酸亚铁起到关键的作用。在没有硫酸亚铁的情况下,锰几乎无法浸出。这是因为软锰矿不溶于缺少还原剂的硫酸溶液或去离子水[14]。当溶液中存在硫酸亚铁作为还原剂时,Mn4+被还原为Mn2+,进而溶于硫酸溶液。因此,锰还原率随着硫酸亚铁的增加而快速增加。当硫酸亚铁与二氧化锰质量比超过2.08∶1时,锰还原率达到饱和。
2.1.6 pH 值的影响
设定软锰矿粒度为45~52 μm,液固质量比为3∶1,m(硫酸亚铁)∶m(二氧化锰)=2.08∶1,搅拌速度为400 r/min,浸出温度为80℃,浸出时间为2 h。通过调整硫酸亚铁与一氧化锰质量比,研究浆液中pH值对锰浸出的影响,结果见图6。
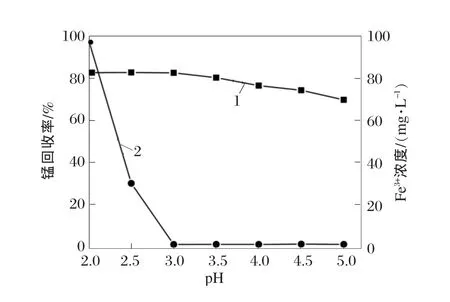
图6 pH值对锰回收率的影响
从图6可以看出:当pH值从5.0降低到3.0时,锰回收率从69%增加到82%,表明较高的硫酸浓度不仅加快浸出率而且有助于提高二氧化锰的还原率,这都是由于溶液中氢离子浓度增加的缘故。随后,Mn2+较为容易的进入溶液中[21]。当pH值从2.0增加到3.0时,浆液中Fe3+浓度迅速从98 mg/L下降到低于1 mg/L。这是因为浆液中在发生方程式(4)的反应,并形成针铁矿。当浆液中pH值超过3.0时,Fe3+浓度基本保持在1 mg/L。因此,在试验中,不需要使用过量的一氧化锰将pH值调至高于3.0。
2.1.7 浸出温度的影响
设定软锰矿粒度为45~52 μm,液固质量比为3∶1,m(硫酸亚铁)∶m(二氧化锰)∶m(一氧化锰)=2.08∶1∶0.24,搅拌速度为 400 r/min。通过调整浸出温度,研究浸出温度在不同浸出时间下对锰浸出的影响,结果见图7。
从图7可以看出:随着浸出温度的升高,锰回收率最大值从 20℃下的 2.6%增加到 80℃下的82.53%,这说明较高的浸出温度可以加速反应速度。由于在浸出温度为90℃时,锰回收率最大值与在80℃时接近,因此,从经济角度考虑,不需要浆液温度在超过90℃的情况下进行浸出。另外,如果在90℃下进行浸出,将导致蒸发更多的水份,影响最终结果。
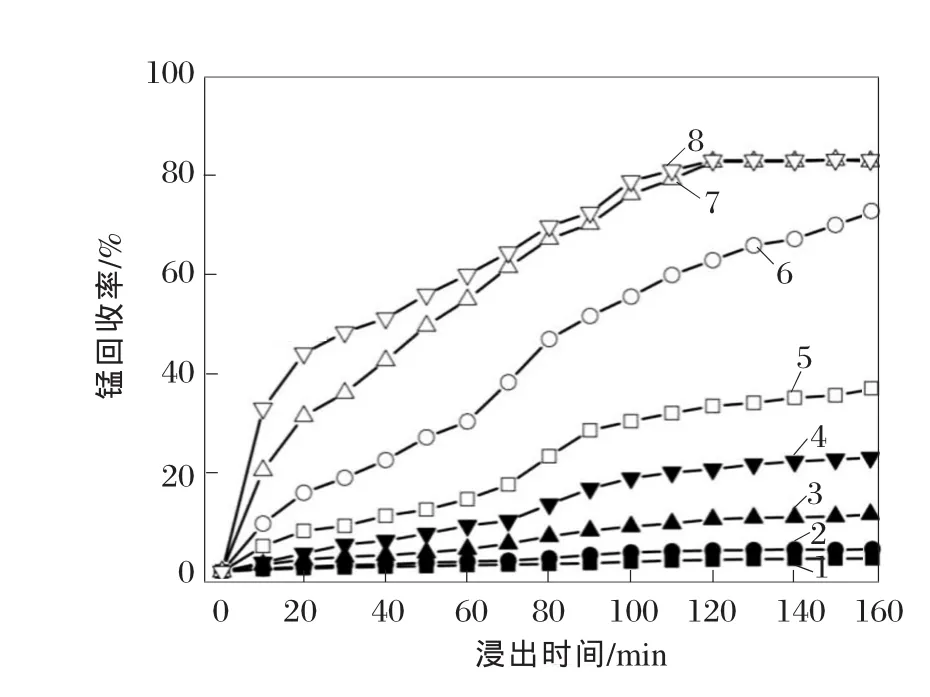
图7 浸出温度在不同浸出时间对锰回收率的影响
为了更全面理解浸出的过程,利用固液反应的动力学模型对图7中的数据进行分析。如果假设软锰矿颗粒成均匀球形固相,那么锰回收率可以利用反应控制的收缩核模型进行分析。以下利用化学反应控制的动力学方程式对试验数据进行检验[22]。

其中,X是参与反应的物质占原物质的比例,t是浸出时间,kc是化学反应速率。
不同浸出温度下,1-(1-X)1/3与浸出时间的曲线如图8所示。直到浸出温度上升到80℃,试验数据都与方程式(6)拟合得较好,而且相关系数都>0.98。这个结果意味着,浸出温度在80℃以下时,在少量硫酸存在的情况下,利用硫酸亚铁从软锰矿中浸出锰的过程是受到表面化学反应所控制的。值得注意的是,在浸出温度为90℃时,试验数据的拟合直线没有经过原点,而其他浸出温度下的拟合直线都经过原点。这表明90℃下的浸出过程需要利用其他的动力学模型来进行分析,这个过程可能涉及到扩散反应[21]。
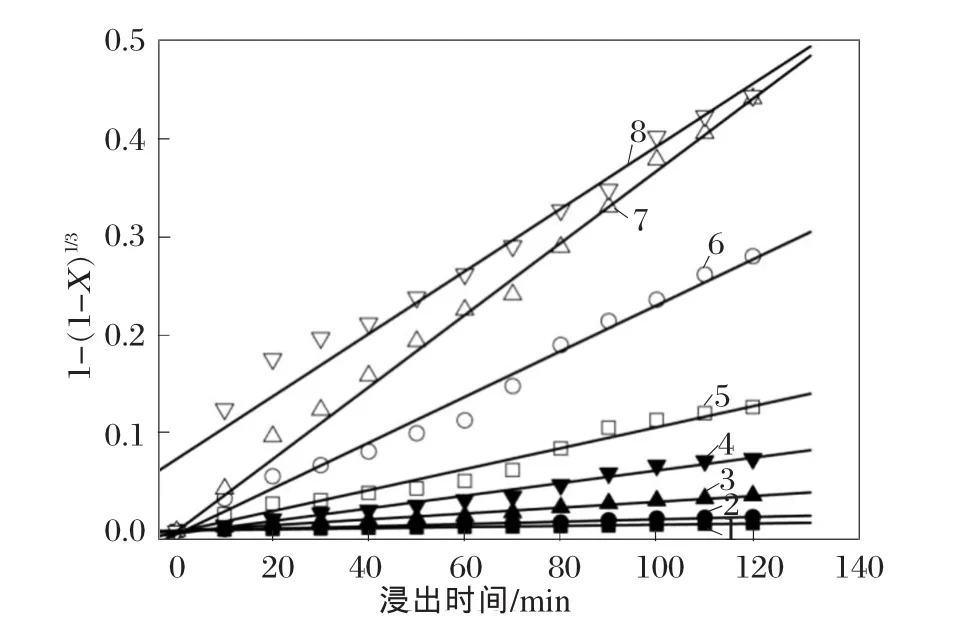
图8 不同温度下的反应动力学关系
经过以上分析,化学方程式(2)的反应活化能可以通过分析反应速率常数与温度的关系曲线获得。这个关系曲线可用以下的阿累尼乌斯方程来表示:

其中,k是反应速率常数,k0是频率因子,Ea是表观活化能,R是气体常数。从图8中,可以计算出不同浸出温度下的反应速率常数。利用该速率常数,可以从lnk与1/T的线性曲线斜率中计算出表观活化能,如图9所示。通过计算,得出方程式(2)的表观活化能为59.6 kJ/mol。
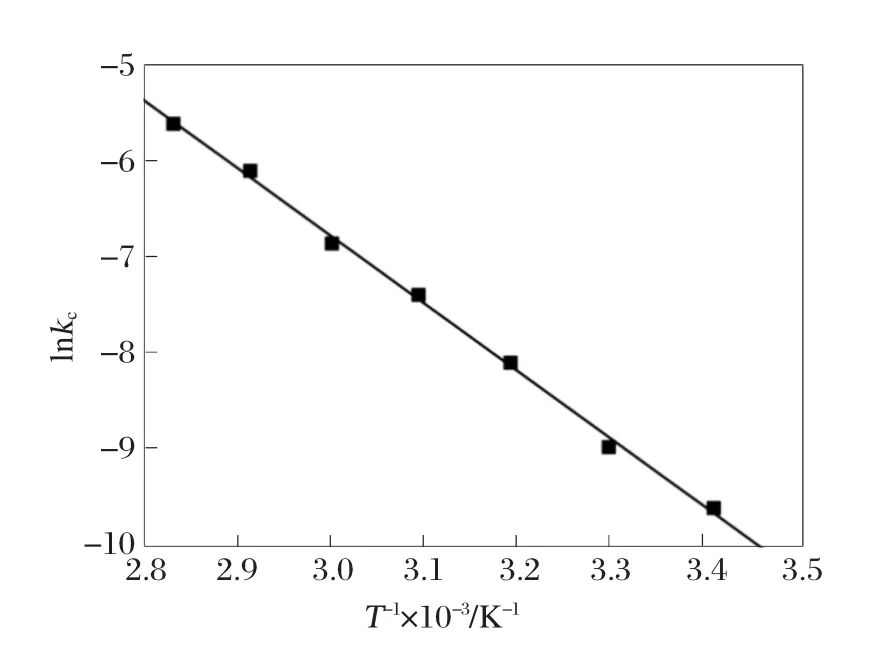
图9 反应速率常数与浸出温度的阿累尼乌斯曲线
2.2 从浸出滤液中回收锰
通过上述试验研究,获得在少量硫酸存在的情况下,利用硫酸亚铁从软锰矿中浸出锰的优化条件,即软锰矿粒度为45~52 μm,液固质量比为3∶1,m(硫酸亚铁)∶m(二氧化锰)∶m(一氧化锰)=2.08∶1∶0.24,搅拌速度为 400 r/min,浸出时间为 2 h,浸出温度为80℃。经过浸出,过滤以及杂质沉淀后,滤液被蒸汽加热至140℃并维持2 h,进行结晶处理。在结晶处理完后,将所得结晶放入离心机进行5 min脱水处理,离心机转速为800 r/min。最后,将脱水结晶进行1 h烘干,烘干温度为150℃。表3所示为经过上述工艺后,所得粉末的化学成分及其特性,其中含有高纯度的一水硫酸锰以及微量杂质。
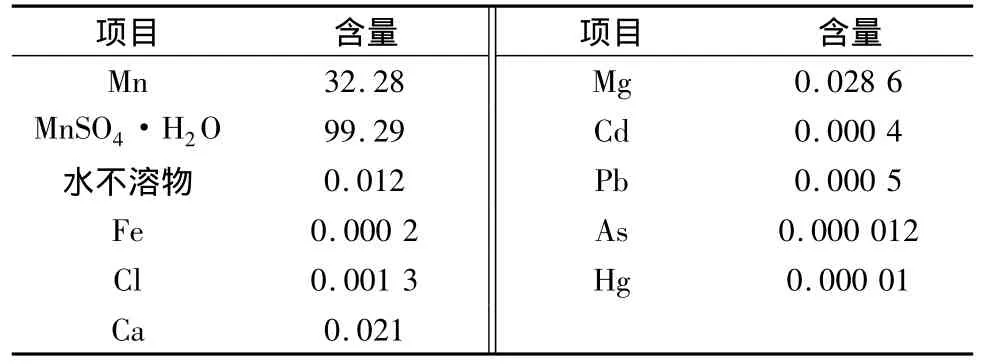
表3 所得粉末的化学成分及含量(质量分数)/%
3 结论
利用钛白副产物硫酸亚铁作为还原剂,对软锰矿的锰还原浸出进行调查研究,分析不同浸出条件对锰回收率的影响。基于各种试验数据,获得优化的浸出条件,即软锰矿粒度为45~52 μm,液固质量比为3∶1,m(硫酸亚铁)∶m(二氧化锰)∶m(一氧化锰)=2.08∶1∶0.24,搅拌速度为 400 r/min,浸出时间为2 h,浸出温度为80℃。所得粉末含99.29%MnSO4·H2O以及微量杂质,这个品质已经超过大多数工业客户的要求。由于钛白副产物在中国价格便宜且产量较大,使得该工艺对于软锰矿的锰浸出来说是一个技术有效且经济可行的方法。
试验数据的动力学分析显示,浸出温度在20~80℃的范围内,浸出过程与化学反应模型可以很好的拟合。通过拟合模型,得到浸出过程的计算活化能为 59.6 kJ/mol。
[2]Elsherief A E.A study of the electroleaching of manganese ore[J].Hydrometallurgy,2000(55):311 -326.
[3]De Michelis I,Ferella F,Beolchini F,et al.Characterisation and classification of solid wastes coming from reductive acid leaching of low-grade manganiferous ore[J].J.Hazard.Mater,2009,162(2-3):1285-1291.
[4]Belyi A V,Pustoshilov P P,Gurevich Y L,et al.Bacterial leaching of manganese ores[J].Appl.Biochem.Microbiol,2006,42(3):289-292.
[5]Misra V N,Khangaonkar P R.Reduction rates of Mn ore pallets with hydrogen[J].Trans.India Inst.Metall.,1975,28(2):169-172.
[6]Guven A,Hurman E.Kinetics of the solid-state carbothermic reduction of wessel manganese ore[J].Metall.Mater.Trans.B,1995(26B):13-24.
[7]Momade F W Y,Momade Z G.A study of the kinetics of reductive leaching of manganese oxide ore in aqueous methanol-sulphuric acid medium[J].Hydrometallurgy,1999(54):25 -39.
[8]Trifoni M,Veglio F,Taglieri G,et al.Acid leaching process by using glucoseas reducingagent:acomparison amongthe efficiency of different kinds of manganiferous ores[J].Miner.Eng.,2000,13(2):217 -221.
[9]Sahoo R N,Naik P K,Das S C.Leaching of manganese from lowgrade manganese ore using oxalic acid as reductant in sulphuric acid solution[J].Hydrometallurgy,2001(62):157 - 163.
[10]Cheng Z,Zhu Guocai,Zhao Yun.Study in reduction-roast leaching manganese from low-grade manganese dioxide ores using cornstalk as reductant[J].Hydrometallurgy,2009(96):176 -179.
[11]Jana R K,Premchard,Pandey B D.Ammoniacal leaching of roast reduced deepsea manganese nodules[J].Hydrometallurgy,1999(53):45-56.
[12]Jiang T,Yang Y,Huang Z,et al.Simultaneous leaching of manganeseand silverfrom manganese-silverores atroom temperature[J].Hydrometallurgy,2003(69):177 -186.
[13]Jiang T,Yang Y,Huang Z,et al.Leaching kinetics of pyrolusite from manganese-silver ores in the presence of hydrogen peroxide[J].Hydrometallurgy,2004(72):129 -138.
[14]Ismail A A,Ali E A,Ibrahim I A,et al.A comparative study on acid leaching of low grade manganese ore using some industrial wastes as reductant[J].Can.J.Chem.Eng.,2004(82):1296-1300.
[15]Brantley F E,Rampacek C.Manganese and iron recovery from leach solutions[P].US:3397130,1968.
[16]Das S C,Sahoo P K,Rao P K.Extraction of manganese from lowgrade manganese ores by ferrous sulfate leaching[J].Hydrometallurgy,1982,8(1):35-47.
[17]Vu H,Jandova J,Lisa K,et al.Leaching of manganese deep ocean nodules in FeSO4-H2SO4-H2O solutions [J].Hydrometallurgy,2005(77):147-153.
[18]Wang D,Song Q,Peng R.Leaching of manganese from low-grade pyrolusite ore with ferrous sulfate and the method of sodium jarosite precipitation[J].J.Northeastern University(Natural Science),1998,19(2):168-170.
[19]Loan M,Newman O M G,Cooper R M G,et al.Defining the Paragoethite process for iron removal in zinc hydrometallurgy[J].Hydrometallurgy,2006(81):104-129.
[20]Song J,Zhu G,Zhang P,et al. Reduction of low-grade manganese oxide ore by biomass roasting[J].Acta Metall.Sin.(Engl.Lett.),2010,23(3):223 -229.
[21]Nayl A A,Ismail I M,Aly H F.Recovery of pure MnSO4·H2O by reductive leaching of manganese from pyrolusite ore by sulfuric acid and hydrogen peroxide[J].Inter.J.Miner.Process.,2011(100):116-123.
[22]Levenspiel O.Chemical reaction engineering[M].NewYork:Wiley,1972:359 -368.
猜你喜欢
农村青少年科学探究(2022年3期)2022-05-13
化学教学(2021年6期)2021-11-17
粉末冶金技术(2021年3期)2021-07-28
小型微型计算机系统(2020年10期)2020-10-21
中国金属通报(2020年20期)2020-03-27
化学教学(2016年12期)2017-07-25
山东工业技术(2016年15期)2016-12-01
浙江大学学报(工学版)(2016年11期)2016-06-05
中南大学学报(自然科学版)(2015年9期)2015-10-13
化学教学(2015年1期)2015-03-19
