
H2在AlnCr(n=1-7)团簇上吸附和解离的密度泛函研究
2015-03-23 05:04李向富李高清
原子与分子物理学报 2015年5期
李向富, 李高清
(陇东学院电气工程学院, 庆阳 745000)
H2在AlnCr(n=1-7)团簇上吸附和解离的密度泛函研究
李向富, 李高清
(陇东学院电气工程学院, 庆阳 745000)
采用密度泛函理论中的B3LYP方法研究了H2在AlnCr(n=1-7)团簇上的吸附和解离.结果表明:AlnCr团簇结构与Aln+1团簇结构相似;物理吸附是H2以侧向的形式吸附在Cr原子上,H-H键长略微增长,H2的振动频率发生了红移;除了n=5外,其它AlnCrH2团簇的最稳定结构均是AlnCr团簇的最稳定结构与两个氢原子成键而成;AlnCr团簇向H原子转移了电荷;AlnCrH2团簇的平均结合能,垂直电离势和能隙均大于AlnCr团簇的,即AlnCrH2团簇比AlnCr团簇更稳定;Al7Cr对H2的化学吸附表现出较强的惰性,而AlnCrH2(n=1,2,6)则表现出较强的化学活性;由化学反应路径跟踪可知,通过改变AlnCr团簇中Al原子的个数可以调节H2的物理化学吸附行为.
AlnCr团簇; 氢分子; 吸附; 解离
1 引 言
2 计算方法
采用用密度泛函理论中的B3LYP方法研究了H2在AlnCr(n=1-7)团簇上的吸附和解离.所有计算均在Gaussian03程序包[12]下完成.Al和H采用全电子基组6-31g*,Cr原子的内层电子和价电子均采用LANL2DZ赝势.根据最小振动频率是否为正值判断所得结构是否为局域稳定结构.使用QST方法寻找物理吸附到化学吸附的中间过渡态,通过计算反应路径来确认过渡态的正确性.为了使得所得结构尽可能是全局最稳定结构,根据已报道的Aln和AlnX(X=Cu, Au, Co, V) 团簇的几何结构,通过替换或担载的方式获得丰富的初始结构.考虑了自旋多重度的影响.
为了验证所采用计算方法的可行性,计算了Cr2, Al2和AlCr二聚体,结果见表1.由表1可以看出:Cr2和Al2的键长、平均结合能和垂直电离势的本文计算值与实验值符合得很好;AlCr的键长和垂直电离势的本文计算值与理论和实验值符合得很好,但是,平均结合能比实验值小得多,而与理论值符合得很好.综上所述,本文采用的计算方法是可行的.
表1 Cr2、Al2和AlCr团簇的键长r、平均结合能Eb、垂直电离势VIP的理论和实验值
Table 1 Theoretical and experimental values of bond length r, average binding energy Eband vertical ionization potential IP of Cr2, Al2and AlCr clusters

Cr2Al2AlCrthisworkExpt.[13]thisworkExpt.[14]thisworkTheo.15]Expt.[16]r(Å)1.6111.6792.5142.5602.7422.742.74Eb(eV)1.5951.420±0.1001.1890.997±0.1081.1251.172.272±0.009IP(eV)6.0996.3046.200±0.2005.9875.945.96±0.04
3 结果与讨论
3.1 AlnCr (n=1-7)团簇的基态结构
图1(na) 给出了AlnCr (n=1-7)团簇的基态结构.表2给出了AlnCr(n=1-7)团簇基态结构的自旋多重度、对称性、总能量、能隙、平均结合能、垂直电离势和垂直电子亲和势.AlCr的键长为2.611 Å,自旋多重度为6,与文献[17]的结果相一致;Al2Cr呈平面等腰三角形,自旋多重度为5,呈C2V对称性;Al3Cr是呈CS对称性的三棱锥结构,自旋多重度为4;Al4Cr是呈C4V对称性的四棱锥结构,自旋多重度为5;Al5Cr是在Al4Cr的四边形底面一侧邻接一Cr原子,自旋多重度为4;Al6Cr是在三棱柱Al6团簇的四边形面上邻接一Cr原子,自旋多重度为5,对称性为C2V;Al7Cr 团簇是在呈C4V对称性的Al6团簇上邻接一Al原子和Cr原子,自旋多重度为6.总之,AlnCr团簇的基态结构均是Cr原子替换Aln+1团簇[18]中一个Al原子而得到.
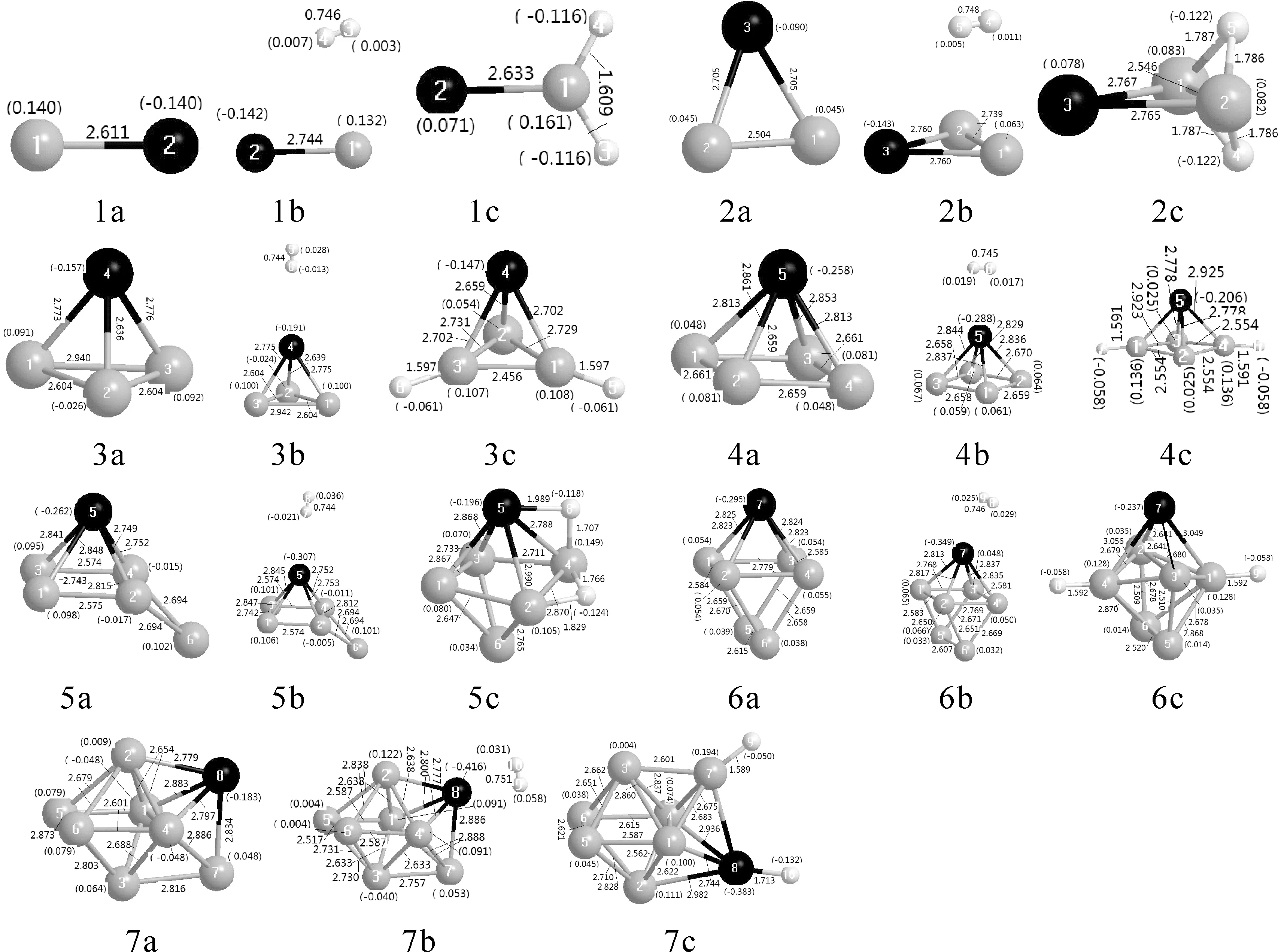
图1 AlnCr (na), AlnCr-H2 (nb)和AlnCrH2 (nc)团簇的基态结构 (灰、黑、白色小球分别表示Al, Cr, H原子)Fig.1 Ground state structures of AlnCr (na), AlnCr-H2 (nb) and AlnCrH2 (nc) clusters (gray ball: Al, black ball: Cr, white ball: H)
表2 AlnCr(n=1-7)团簇基态结构的自旋多重度M,对称性Sym,总能量E,能隙Gap,平均结合能Eb,垂直电离势VIP和垂直电子亲和势VEA
Table 2 Multiplicities M, symmetries Sym, total energies Et, HOMO-LUMO Gaps Gap, averaged binding energies Eb, Vertical ionization potentials VIP and Vertical electron affinities VEA for the most stable AlnCr(n=1-7) clusters

ClusterMSymEt(eV)Gap(eV)Eb(eV)VIP(eV)VEA(eV)AlCr6C∞V-8943.8141.9051.1245.9950.605Al2Cr5C2V-15540.7011.3861.3946.1981.441Al3Cr4CS-22138.0161.2371.6275.9991.312Al4Cr5C4V-28735.2201.2651.7136.1441.793Al5Cr4C1-35332.0181.1341.6845.8991.902Al6Cr5C2V-41930.1471.2601.8876.2812.159Al7Cr6C1-48527.631271.4201.9395.8921.746
3.2 H2在AlnCr (n=1-7)团簇上的物理吸附
考虑了顶位,桥位和面位三种可能的吸附模式.最稳定的吸附结构如图1(nb)所示.吸附能,能隙,H-H键长,平均H-Cr键长和H-H振动频率如表3所示.吸附能的定义式如下:
ΔEad=E(AlnCr)+E(H2)-E(AlnCr-H2)
(1)
(1)式中E(AlnCr ), E(H2)和E(AlnCr -H2)分别表示AlnCr,自由H2和AlnCr-H2团簇的能量.吸附能均为负值,即都是放热反应.H2分子以侧向的形式吸附在Cr原子的顶位上,这是由于Cr原子低配位,化学键未饱和所致.H-H键长范围为0.744—0.751 Å,相对自由H2键长0.7428 Å,略微变长;吸附能的范围为0.009-0.064 eV,相对较小;两个H原子与Cr原子的平均距离为2.391-3.701 Å,是典型的范德华相互作用;这三点均说明了典型的物理吸附特征.吸附在团簇上氢分子的振动频率为4295-4427 cm-1,相对自由H2的频率4650 cm-1,发生了红移,这是由氢分子键长增加所致.
表3 H2物理吸附在AlnCr(n=1-7)团簇上的吸附能△Ead,能隙Gap,H-H键长DH-H,平均H-Cr键长DH-Cr和H-H振动频率ωH-H
Table 3 Adsorption energies △Ead, HOMO-LUMO gaps Gap, H-H bond lengths DH-H, averaged H-Cr bond lengths DH-Crand H-H vibration frequencies ω for the physisorption of H2on AlnCr(n=1-7) clusters
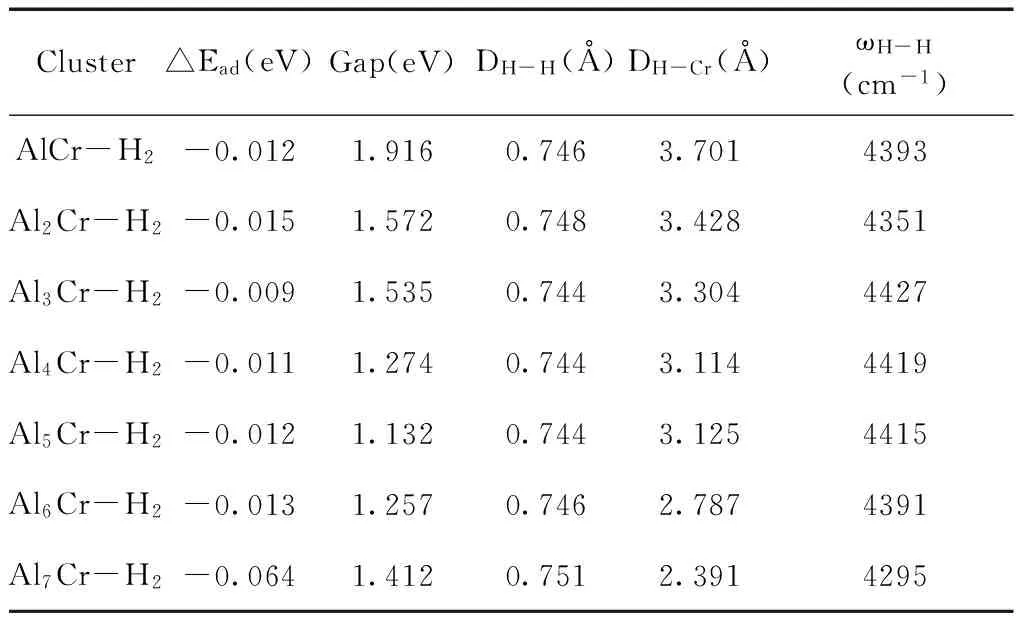
Cluster△Ead(eV)Gap(eV)DH-H(Å)DH-Cr(Å)ωH-H(cm-1)AlCr-H2-0.0121.9160.7463.7014393Al2Cr-H2-0.0151.5720.7483.4284351Al3Cr-H2-0.0091.5350.7443.3044427Al4Cr-H2-0.0111.2740.7443.1144419Al5Cr-H2-0.0121.1320.7443.1254415Al6Cr-H2-0.0131.2570.7462.7874391Al7Cr-H2-0.0641.4120.7512.3914295
3.3 H2在AlnCr (n=1-7)团簇上的化学吸附
AlnCrH2的最稳定吸附结构如图1(nc)所示.AlCrH2是平面结构,H-Al键长均为1.609 Å,H原子获得等量电荷(0.116e).Al2CrH2是两个氢原子分别邻接在Al-Al键桥位上,H-Al键长几乎相等(1.786 Å,1.787 Å),H原子获得等量电荷(0.122e).Al3CrH2是两个氢原子分别邻接在两个Al原子的顶位上,H-Al键长均为1.597 Å,H原子获得等量电荷(0.061e).Al4CrH2是两个氢原子分别邻接在相对的两个Al原子的顶位上,H-Al键长均为1.591 Å,H原子获得等量电荷(0.058e).Al5CrH2的基态结构相对Al5Cr发生了根本性变化,不是在Al5Cr基础上邻接H原子,而是在Al5Cr亚稳定结构基础上邻接H原子,一个H位于Al-Al键桥位上,另一个H位于Al-Cr桥位上,两个H原子得到电荷分别为0.118e和0.124e.Al6CrH2是两个H原子分别接在相对的两个Al原子的顶位上,H-Al键长均为1.592 Å,H原子获得等量电荷(0.058e).Al7CrH2团簇中一个H原子接在Al的顶位上,一个H原子接在Cr的顶位上;H-Al键长为1.589 Å,H-Cr键长为1.713 Å;H分别获得电荷(0.056e, 0.132e).总之,n=5的最稳定结构不是由Al5Cr的最稳定结构吸附两个H原子,其它AlnCrH2的最稳定结构均是在最稳定的AlnCr团簇上吸附两个氢原子;AlnCr向H原子转移了电荷.
由表4和图1(nc)可以看出,AlnCrH2团簇中H-H距离远远大于H2分子中的H-H键长,即H2发生了解离化学吸附.化学吸附能大小反应了H2与团簇间的化学反应活性,其定义式如下:
ΔECE=E(AlnCr)+E(H2)-E(AlnCrH2)
(2)
(2)式中E(AlnCr ),E(H2),E(AlnCrH2)分别表示AlnCr团簇,自由H2和AlnCrH2团簇的能量.由表4可以看出Al7CrH2的化学吸附能最小,即Al7Cr与H2相互作用时表现出较强的惰性;而AlCrH2,Al2CrH2和Al6CrH2的化学吸附能相对较大,对H2的化学吸附表现出较强的化学活性.
表4 AlnCrH2团簇的吸附能△ECE、平均结合能Eb、能隙Gap、H-H距离DH-H、垂直电离势VIP和垂直亲和势VEA
Table 4 Adsorption energies △ECE, average binding energies Eb, HOMO-LUMO gaps, distances between H-H DH-H, Vertical ionization potentials VIP and Vertical electron affinities VEA for the most stable AlnCrH2(n=1-7) clusters
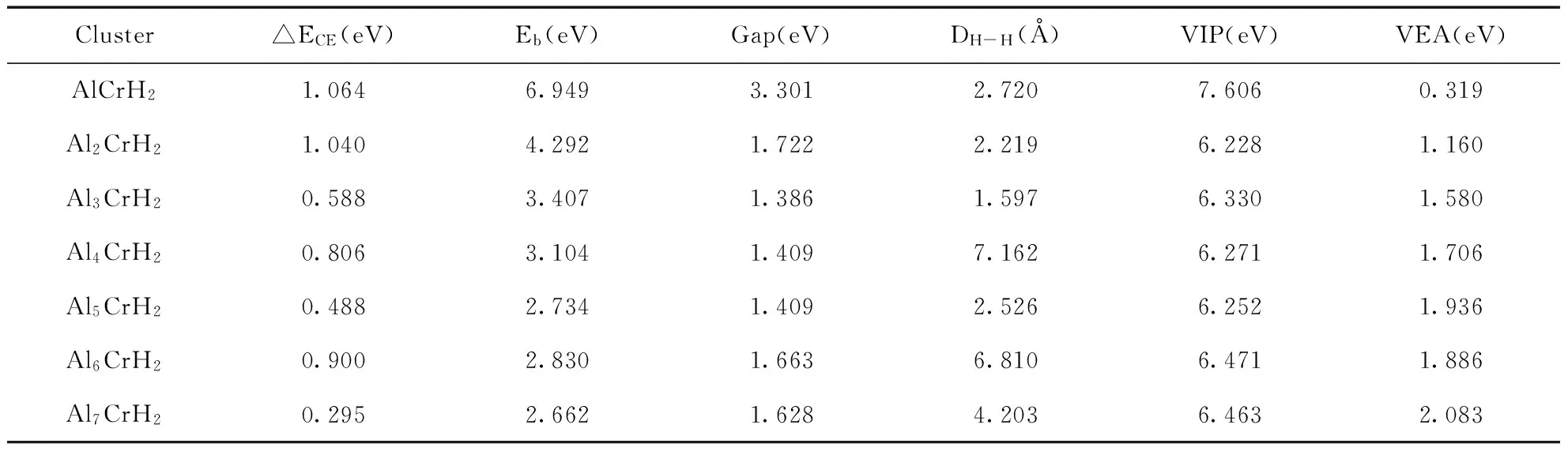
Cluster△ECE(eV)Eb(eV)Gap(eV)DH-H(Å)VIP(eV)VEA(eV)AlCrH21.0646.9493.3012.7207.6060.319Al2CrH21.0404.2921.7222.2196.2281.160Al3CrH20.5883.4071.3861.5976.3301.580Al4CrH20.8063.1041.4097.1626.2711.706Al5CrH20.4882.7341.4092.5266.2521.936Al6CrH20.9002.8301.6636.8106.4711.886Al7CrH20.2952.6621.6284.2036.4632.083
3.4 AlnCr与AlnCrH2的稳定性比较
通过计算平均结合能、能隙、垂直电离势和垂直亲和势比较了AlnCr与AlnCrH2的稳定性.平均结合能的定义式如下:
Eb(AlnCr)=[nE(Al)+E(Cr)-E(AlnCr)]/n
(3)
Eb(AlnCrH2)=[nE(Al)+E(Cr)+
2E(H)-E(AlnCrH2)]/n
(4)
由表2,表4和图2可以看出,AlnCr 和AlnCrH2团簇的平均结合能变化范围分别为:1.124-1.939 eV,2.662-6.949 eV,很明显AlnCrH2团簇的平均结合能大于AlnCr团簇,即AlnCrH2团簇更稳定;随着团簇尺寸的增加,AlnCrH2团簇的平均结合能逐渐减小,AlnCr团簇的平均结合能逐渐增加;可以推测出当团簇尺寸大于某一临界值时,二者的平均结合能几乎相等,因为那时H2分子的影响可以忽略了.
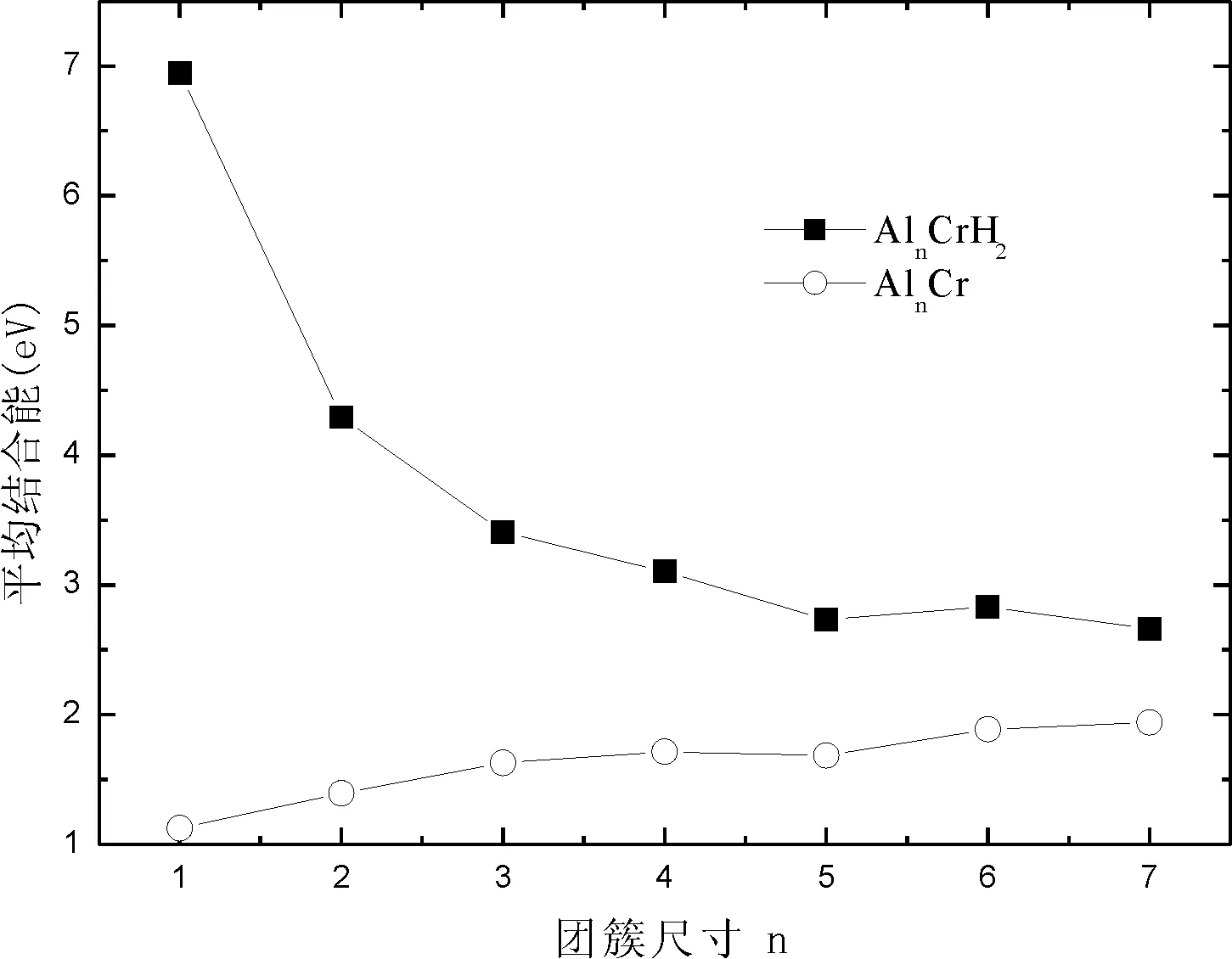
图2 AlnCr和AlnCrH2团簇平均结合能随团簇尺寸的变化Fig.2 Average binding energies per atom of AlnCr and AlnCrH2 versus cluster size
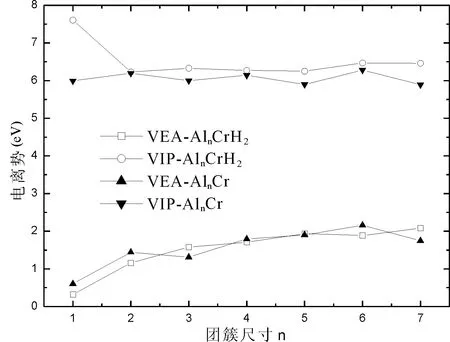
图3 AlnCr和AlnCrH2团簇垂直电离势和垂直亲和势随团簇尺寸的变化Fig.3 Vertical ionization potentials and Vertical electron affinities of AlnCr and AlnCrH2 versus cluster size
保持阴阳离子团簇结构与中性团簇结构相同的情况下,计算的电离势或亲和势称为垂直电离势或垂直亲和势.由图3可以看出AlnCrH2的电离势均大于AlnCr团簇的电离势,这也说明AlnCrH2团簇更加稳定,较不容易电离电子;AlnCrH2和AlnCr的电子亲和势随团簇尺寸变化趋势相似,表现出奇偶振动现象.由图4可以看出AlnCrH2和AlnCr的能隙随团簇尺寸变化趋势相同,且AlnCrH2的能隙值均大于AlnCr,即AlnCrH2比AlnCr更稳定.
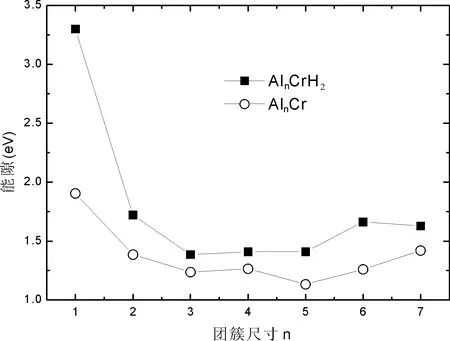
图4 AlnCr和AlnCrH2团簇的能隙随团簇尺寸的变化Fig.4 Energy gaps of AlnCr and AlnCrH2 versus cluster size
3.5 H2分子在AlnCr (n=1-7)团簇上的解离机制
以自由AlnCr和H2的能量之和作为势能零点,以此得出反应物(H2在AlnCr团簇上的物理吸附),过渡态和产物(H2在AlnCr团簇上的化学吸附)的相对能量.反应物、过渡态和产物的能量,反应能(产物与反应物的能量之差)以及反应势垒(过渡态与反应物能量之差)的计算结果见表5.反应能均为负值,说明H2的解离为放热过程.AlCrH2、Al2CrH2和Al6CrH2的反应势垒较小,反应能较大,化学吸附能也较大(见表4);说明AlCrH2、Al2CrH2和Al6CrH2较容易与H2发生化学反应,当H2与它们相互作用时,H-H键较容易断裂.Al7CrH2团簇有最大的反应势垒(17.154 kcal/mol),H2不容易发生化学吸附.详细分析了H2在AlnCr团簇上的解离反应路径,即H2+AlnCr→AlnCrH2,搜寻了中间产物和过渡态.图5给出了H2在AlnCr团簇上的解离反应路径以及反应物,过渡态(只有一个虚频,其值见表5)和产物的几何结构.对于AlnCr(n=1-7,n≠4)团簇,H2由物理吸附到化学吸附经历了一个过渡态(TS),跨越的势垒值分别为:9.507, 8.070, 5.103, 11.989, 0.821, 17.154 kcal/mol;对于Al4Cr团簇,H2由物理吸附到化学吸附经历了两个过渡态(TS1和TS2),首先由反应物R经过渡态TS1生产中间产物IM,跨越的势垒值为27.928 kcal/mol,中间产物H-H间距离为1.242 Å(产物中H-H距离值见表4),H-H键已断裂,两个氢原子分别与相邻的两个Al原子成键;然后由中间态IM反应至最后产物P,跨越的势垒值为11.542 kcal/mol,产物P中的两个H原子处在相对的两个Al原子上,产物P的能量比中间产物IM低了3.733 kcal/mol.上述结果表明,H2在不同尺寸AlnCr团簇上的反应路径是不同的,故可通过改变AlnCr团簇中Al原子的个数来调节H2的物理化学吸附行为.

(a)AlCrH2
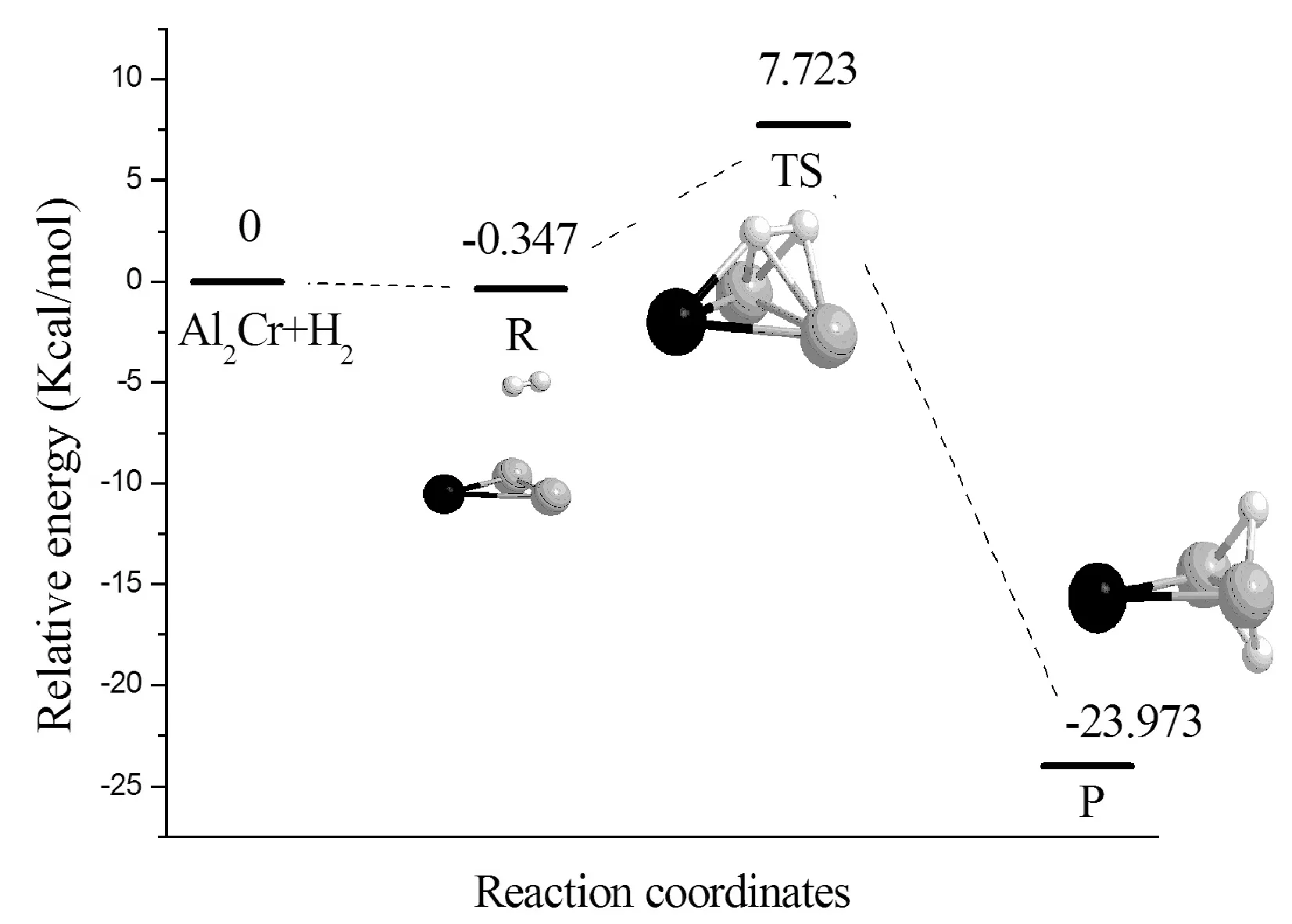
(b)Al2CrH2

(c)Al3CrH2

(d)Al4CrH2
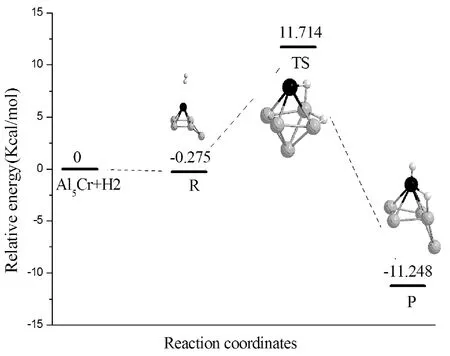
(e) Al5CrH2

(f) Al6CrH2

(g) Al7CrH2图5 H2分子在AlnCr团簇上的解离反应路径Fig.5 The dissociation reaction pathway of H2 on AlnCr clusters
表5 H2分子在AlnCr团簇上解离过程中的反应物能量ER、过渡态能量ETS、产物能量EP、反应能ERe、反应势垒EAc、过渡态的虚频ωTS
Table 5 Energies of reactant ER, energies of transition state ETS, energis of product EP, reaction energies ERe, activation barriers EAcand imaginary frequencies of transition state ωTSfor H2dissociation process on AlnCr clusters
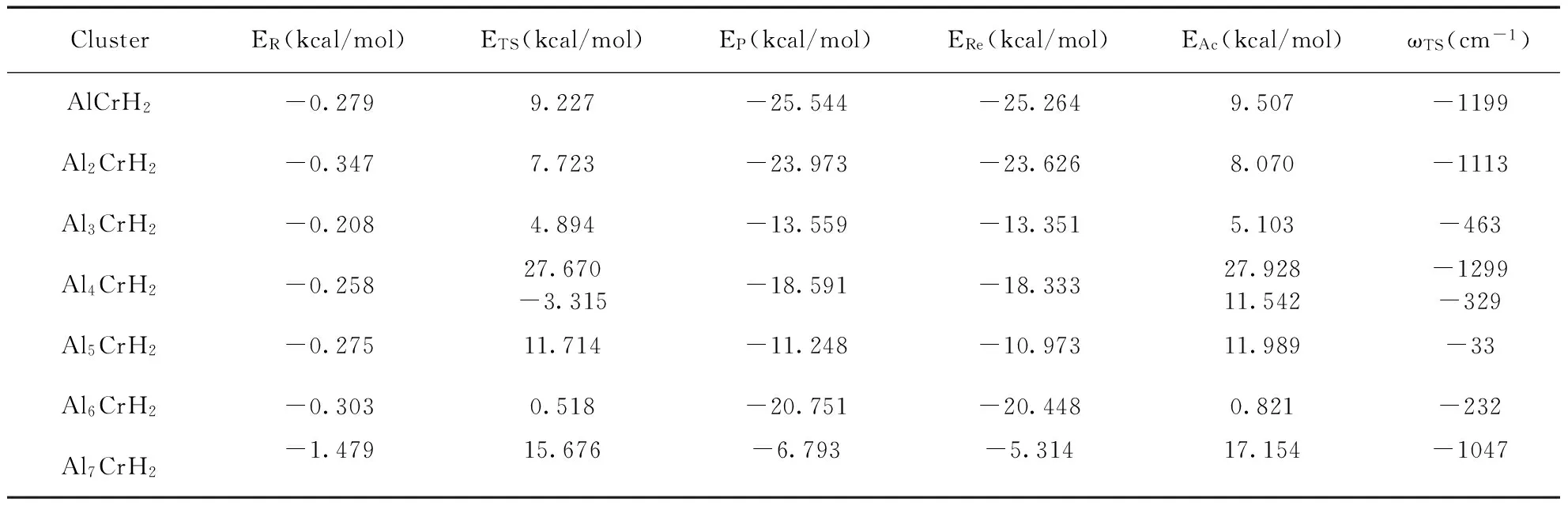
ClusterER(kcal/mol)ETS(kcal/mol)EP(kcal/mol)ERe(kcal/mol)EAc(kcal/mol)ωTS(cm-1)AlCrH2-0.2799.227-25.544-25.2649.507-1199Al2CrH2-0.3477.723-23.973-23.6268.070-1113Al3CrH2-0.2084.894-13.559-13.3515.103-463Al4CrH2-0.25827.670-3.315-18.591-18.33327.92811.542-1299-329Al5CrH2-0.27511.714-11.248-10.97311.989-33Al6CrH2-0.3030.518-20.751-20.4480.821-232Al7CrH2-1.47915.676-6.793-5.31417.154-1047
4 结 论
采用密度泛函理论中的B3LYP方法研究了H2在AlnCr(n=1-7)团簇上的吸附和解离.结果表明:AlnCr团簇结构与Aln+1团簇结构相似;物理吸附是H2以侧向的形式吸附在Cr原子的顶位上,H-H键略微增长,H2的振动频率发生了红移;n=5的最稳定结构不是Al5Cr的最稳定结构吸附两个H原子,其它AlnCrH2的最稳定结构均是在最稳定的AlnCr团簇上吸附两个氢原子;AlnCr向氢原子转移了电荷;AlnCrH2的平均结合能,垂直电离势和能隙均大于AlnCr的,即AlnCrH2比AlnCr更稳定;Al7Cr对H2的化学吸附能最小,表现出较强的惰性,而AlnCrH2(n=1,2,6)对H2的化学吸附能较大,表现出较强的化学活性;H2在不同尺寸AlnCr团簇上的反应路径是不同的,故可通过改变AlnCr团簇中Al原子的个数来调节H2的物理化学吸附行为.
[1] Arakawa H, Aresta M, Armor J N,etal. Catalysis research of relevance to carbon management: progress, challenges, and opportunities[J].Chem.Rev., 2001, 101: 953.
[2] Guo L, Li S Y, Zhang X,etal. Hydrogen adsorption and dissociation on small AlnAu clusters: an electronic structure density functional study[J].Eur.Phys.J. D, 2013, 67: 137
[3] Li X F, Li G Q. Density functional theory study on the stability and electronic properties of Cun(n=1-20) clusters[J].J.At.Mol.Phys., 2014, 31(4): 575 [李向富, 李高清. Cun(n=1-20)团簇的稳定性和电子性质的密度泛函研究[J]. 原子与分子物理学报, 2014, 31(4): 575]
[4] Fayet P, Kaldor A, Cox D M. Palladium clusters: H2, D2, N2, CH4, CD4, C2H4, and C2H6reactivity and D2saturation studies[J].J.Chem.Phys., 1990, 92: 254.
[5] Doyle A M, Shaikhutdinov S K , Jackson S D,etal. Hydrogenation on metal surfaces: Why are nanoparticles more active than single crystals?[J].J.Angew.Chem.Int.Ed., 2003, 42: 5240.
[6] Lee H W, Chang C M. Size effect of Pd clusters on hydrogen adsorption[J].J.Phys.:Condens.Matter, 2011, 23: 045503.
[7] Kadioglu Y, Demirkiran A, Yaraneri H,etal. Investigation of NH3and H2adsorption on Ptn(n=2-15, 18, 22, 24) clusters by using density functional theory[J].JournalofAlloysandCompounds, 2014, 591: 188.
[8] Ge G X, Yan H X, Jing Q,etal. Theoretical study of hydrogen adsorption on ruthenium clusters[J].J.Clust.Sci., 2011, 22: 473.
[9] Ge G X, Cao H B, Jing Q,etal. Density functional theory study of the interaction of H2with rhodium clusters[J].ActaPhysicaSinica, 2009, 58: 8236 (in Chinese)[葛桂贤, 曹海滨, 井群, 等. 密度泛函理论研究H2与Rhn(n=1-8)团簇的相互作用[J]. 物理学报, 2009, 58: 8236]
[10] Guo L, Yang Y F. Theoretical investigation of molecular hydrogen adsorption and dissociation on AlnV(n=1-13) clusters[J].InternationalJournalofHydrogenEnergy, 2013, 38: 3640.
[11] Lu Q L, Wan J G. Sc-coated Si@Al12as high-capacity hydrogen storage medium[J].J.Chem.Phys., 2010, 132: 224308.
[12] Frisch M J, Trucks G W, Schlegel H B,etal. Gaussian 03(revision C02), Pittsburgh, PA: Gaussian, Inc, 2003.
[13] Bondybey V E, English J H. Electronic structure and vibrational frequency of Cr2[J].Chem.Phys.Lett., 1983, 94: 443.
[14] Rosen B.Spectroscopicdatarelativetodiatomicmolecules[M]. New York: Oxford University Press, 1970.
[15] Ouyang Y, Wang J, Liu F,etal. Density functional study of 3d-transition metal Aluminides[J].J.Mol.Struct.:THEOCHEM, 2009, 905: 106.
[16] Behm J M, Brugh D J, Morse M D. Spectroscopic analysis of the open 3d subshell transition metal aluminides: AlV, AlCr, and AlCo[J].J.Chem.Phys., 1994, 101: 6487.
[17] Wang M, Huang X, Du Z,etal. Structural, electronic, and magnetic properties of a series of Aluminum clusters doped with various transition metals[J].Chem.Phys.Lett., 2009, 480: 258.
[18] Kiohara Valéria O, Carvalho Edson F V, Paschoal Carlos W A,etal. DFT and CCSD(T) electronic properties and structures of aluminum cluster: Alxn(n=1-9, x=0,±1)[J].Chem.Phys.Lett., 2013, 568: 42.
Density functional theoretical investigation for the adsorption and dissociation of molecular hydrogen on AlnCr(n=1-7) clusters
LI Xiang-Fu, LI Gao-Qing
(College of Electrical Engineering, Longdong University, Qingyang 745000, China)
The adsorption and dissociation of molecular hydrogen on AlnCr(n=1-7) clusters are investigated by using the method of B3LYP in density functional theory. The results show that the structures of AlnCr clusters are similar to those of Aln+1clusters. H2is easily adsorbed physically on Cr atom with a side-on orientation. The bond length of H-H increases slightly. The vibration frequency of H2adsorbed is smaller than that of free molecular H2, namely, a red-shift occurs. Except forn=5, the most stable structures of the other AlnCrH2clusters are composed by the most stable structures of AlnCr clusters and two H atoms. Charge transfers from AlnCr clusters to H atoms. Average binding energy, vertical ionization potential and energy gap of AlnCrH2clusters are all greater than those of AlnCr clusters, namely AlnCrH2clusters are more stable than AlnCr clusters. Al7Cr cluster shows stronger inertness for chemical adsorption of H2, while AlnCrH2(n=1,2,6) show stronger chemical activity. The physical and chemical adsorption behavior of H2on AlnCr clusters can be adjusted by changing the number of Al atom in AlnCr clusters from tracking the chemical reaction path.
AlnCr clusters; Molecular hydrogen; Adsorption; Dissociation
李向富(1982—),男,甘肃环县人,理学硕士,主要从事团簇结构及性质的研究.
李高清. E-mail: lgaoq@163.com
103969/j.issn.1000-0364.2015.10.010
O641
A
1000-0364(2015)05-0775-08
投稿日期:2015-03-21
猜你喜欢
大学物理(2022年9期)2022-09-28
北京航空航天大学学报(2022年5期)2022-06-06
物理通报(2020年7期)2020-07-01
青岛大学学报(工程技术版)(2019年2期)2019-09-10
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
火炸药学报(2012年4期)2012-01-29
