
2-氯-5-三氟甲基苯胺水杨醛Schiff碱的结构及光谱性质研究
2015-03-23 04:04孟德素卢金凤
原子与分子物理学报 2015年5期
孟德素, 卢金凤
(菏泽学院化学化工系, 菏泽 274015)
2-氯-5-三氟甲基苯胺水杨醛Schiff碱的结构及光谱性质研究
孟德素, 卢金凤
(菏泽学院化学化工系, 菏泽 274015)
实验以2-氯-5-三氟甲基苯胺、水杨醛为原料,合成2-氯-5-三氟甲基苯胺水杨醛Schiff碱,测定了Schiff碱的红外光谱和氢谱.利用密度泛函理论(DFT)B3LYP方法在6-311++G**基组上对合成的Schiff碱化合物进行了几何构型的优化和红外光谱的计算,得到了分子的优势构象、频率值及对应的红外强度,并比较了实验和理论计算的光谱数据,对分子的振动模式进行了全面的光谱归属,发现理论计算与实验测试结果吻合的较好.
2-氯-5-三氟甲基苯胺; 水杨醛; Schiff碱; DFT; 振动光谱
1 引 言
Schiff碱是一类非常重要的含氮配体,它是由醛或酮的羰基与伯胺、肼及其衍生物的NH2缩合而得,是一种有碳氮双键连结形成的化合物.Schiff碱在合成上具有很大的灵活性,能跟金属离子很好的络合,同时Schiff碱类化合物具有一定的生理学和药理学活性,使得Schiff碱及其配合物的研究十分广泛,特别是在其合成、结构及应用等方面[1],Schiff碱具有抗菌[2]、抗肿瘤[3]、与DNA相互作用等生物活性[4],引起了科学界的广泛关注.因此,研究Schiff碱类化合物的结构和振动光谱,准确地掌握其变化规律有重要的意义[5].目前常用的量子化学方法中的密度泛函理论可以处理相关电子的问题,计算分子结构和红外振动光谱等参数,并与实验数据吻合的很好[6].近年来,量子化学中的密度泛函理论在分子的拉曼光谱与红外振动光谱上得到广泛的应用研究,在与大量的实验结果对比中显示了它的准确性[7].
实验合成了2-氯-5-三氟甲基苯胺Schiff碱,用红外光谱和氢谱对其进行了结构表征,利用密度泛函理论B3LYP方法在6-311++G**基组上优化出了分子可能存在的构象,找出稳定的构象,通过实验和理论数据的结合,对红外光谱进行了指认,得到预测Schiff碱类化合物结构和红外光谱的理论方法,为进一步研究Schiff碱类化合物提供了基础.
反应路线如下:
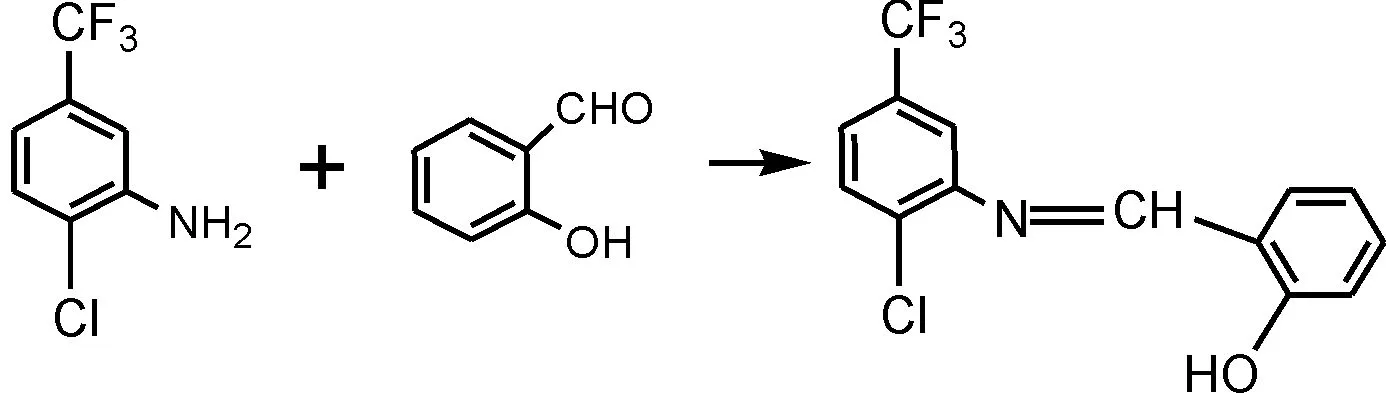
2 2-氯-5-三氟甲基苯胺水杨醛Schiff碱的合成
2.1 仪器与试剂
FTIR-370傅里叶红外光谱仪(美国尼高利公司);MP-21型显微熔点仪(上海精密仪器仪表有限公司);500MHz超导核磁共振仪(瑞士布鲁克公司).
水杨醛(武汉有机实业股份有限公司);2-氯-5-三氟甲基苯胺(上海璞光实业有限公司);无水乙醇(天津市河东区红岩试剂厂).
2.2 Schiff碱的合成与表征
2-氯-5-三氟甲基苯胺Schiff碱的合成及红外光谱和氢谱的表征已在2-氯-5-三氟甲基苯胺及其Schiff碱的合成与表征[8]一文中表述,用FTIR-370傅里叶红外光谱仪,以KBr压片法,在400~4000 cm-1内扫描,得到2-氯-5-三氟甲基苯胺水杨醛Schiff碱的红外光谱图,见图1.
由图1可见,2-氯-5-三氟甲基苯胺水杨醛Schiff碱在1573.43、1481.31、1454.59 cm-1处的一组吸收峰属于芳环骨架的伸缩振动,3440.30 cm-1为OH的伸缩振动,在1621.01 cm-1处有强吸收峰,为C=N双键的伸缩振动峰,表明亚氨基的存在,从而证明Schiff碱的形成.
3 计算方法
2-氯-5-三氟甲基苯胺水杨醛Schiff碱分子利用密度泛函理论的方法在B3LYP/6-311++ G**理论水平上进行了几何构型的优化,得到了其能量及键长、键角和二面角等结构参数,在优化结构的基础上计算了该分子的红外光谱振动频率,并对振动模式进行了分析和归属.
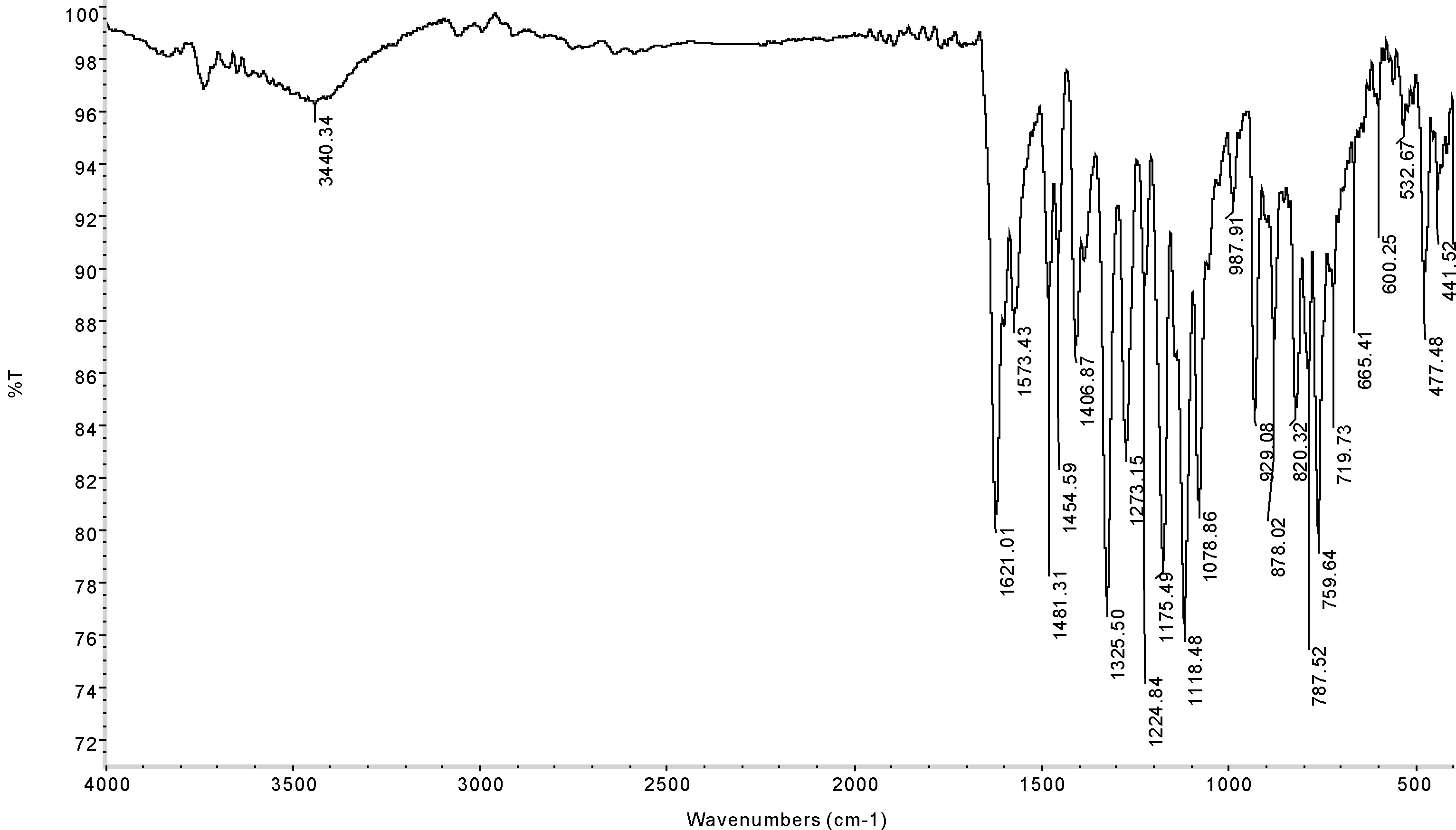
图 1 schiff碱的实验红外光谱图Fig. 1 Experimental infrared spectrum of schiff base
4 结果与讨论
4.1 分子的几何构型
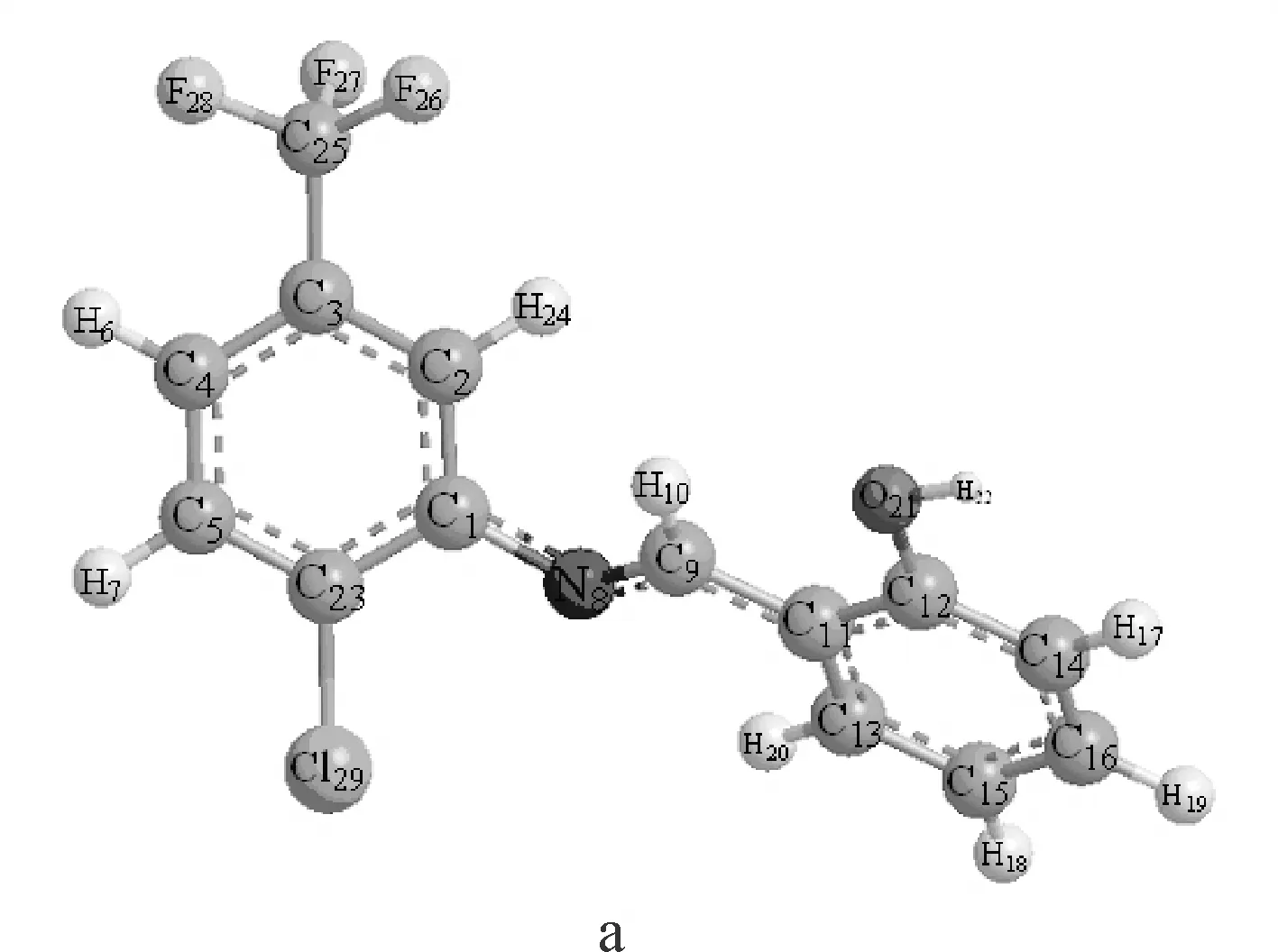
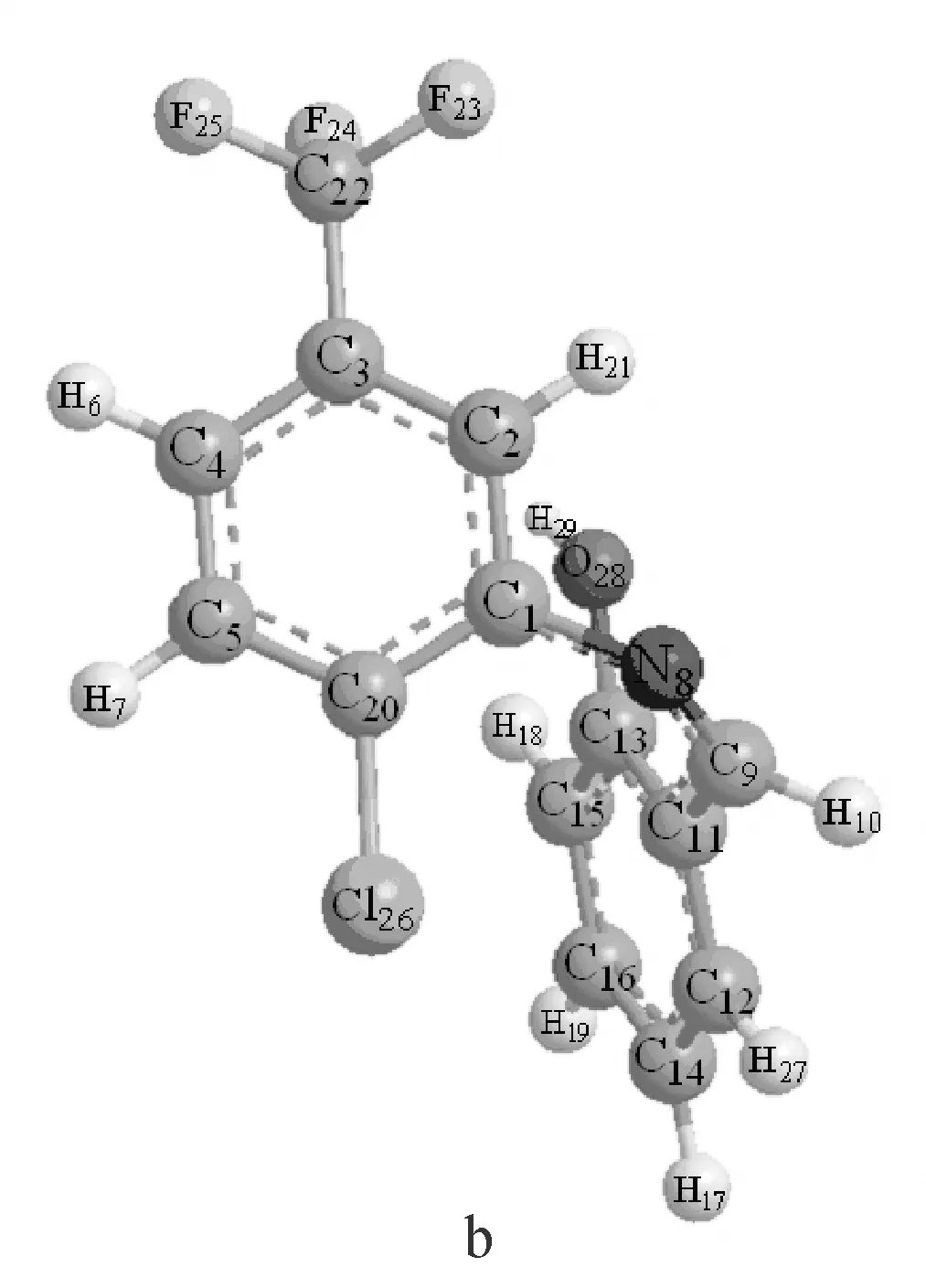
图2 用B3LYP/6-311++G**理论优化出的Schiff碱的优势构象a和bFig.2 Optimized molecular structure a and b of Schiff base at B3LYP/6-311++G** level

构象能量/au点群偶极矩/debyea-1428.90972211C14.4920b-1428.89650710C14.8859
在优化出的构象里,a构象能量比b构象能量低,能量越低结构越稳定.所以,构象a为2-氯-5-三氟甲基苯胺水杨醛Schiff碱分子最稳定的结构.以下均是对schiff碱稳定构象a的讨论.
由表2可见:2-氯-5-三氟甲基苯胺水杨醛Schiff碱分子中的重原子都采取了sp2杂化方式.由图2和表2可以明显的看出,该分子中两个苯环不在同一平面上,两者之间存在一定的二面角,除D(2,1,8,9)和D(23,1,8,9)两个角度数分别为46.679°和137°以外,其余二面角的绝对值均近似为180°或0°,表明C11-C13-C15-C16-C14-C12苯环与羟基和-C=N-基团在同一平面上,两个苯环之间存在着绝对值为46.679°的二面角,从键长的角度分析:2-氯-5-三氟甲基苯胺水杨醛Schiff碱中的苯环r(11,13)=1.404 Å, r(11,12)=1.408 Å,比单个苯环中C-C键的键长1.40 Å要长;而r(13,15)=1.386 Å,r(14,16)=1.390 Å,比单个苯环中C-C键的键长1.40 Å要短,r(8,9)=1.280 Å比正常的C=N双键的键长1.27 Å要长,r(1,8)=1.395 Å,比正常的C-N单键的键长1.470 Å要短,从这些数据中不难看出,2-氯-5-三氟甲基苯胺水杨醛Schiff碱分子在优化后双键键长变长,单键键长变短,说明了该分子中存在着较大的共轭体系.
4.2 振动频率分析
在优化Schiff碱结构的基础上进行了振动频率分析,计算得到的分子稳定构型的振动光谱数据,实验测定的光谱数据及对应的振动模式的全面指认与归属见表3,考虑到B3LYP的系统误差,修正理论计算值,引入了矫正因子,当频率在1700 cm-1以上使用矫正因子0.96,小于1700 cm-1的频率使用矫正因子0.98[9].
通过模拟单个分子的振动模式理论分析及结合实验测量数据结果和有关资料,对2-氯-5-三氟甲基苯胺水杨醛Schiff碱分子的振动模式进行了详细归属:在红外光谱中的O-H伸缩振动谱带强,O-H伸缩振动的特点是特征峰出现在约3700-3500 cm-1以内(峰尖、强)[10],缔和的OH在3500-3200 cm-1以内峰形强而宽[11].对于Schiff碱分子中实验测定值3440.30 cm-1被指认为OH的伸缩振动,而理论计算值在3680.6 cm-1处,这是由于OH具有很强的极性,分子之间具有极性吸引力产生缔合,实验检测分子中的OH是缔和状态,而理论计算的是单个分子中游离OH的伸缩振动,所以引起频率的较大位移.O-H的面内弯曲振动吸收带在1500~1300 cm-1附近[12],理论计算值在1329.62 cm-1处,与实验测定1325.50 cm-1基本相吻合.
芳烃C-H伸缩振动通常在3100-3000 cm-1处.这是鉴定C-H伸缩特性的区域振动.用FT-IR测得对应Schiff碱苯环的C-H伸缩振动为3056.10 cm-1,与理论计算值3062.69 cm-1基本吻合,2911.33 cm-1在实验中被归属于N=C-H伸缩振动,对应于理论计算值2943.79 cm-1.取代苯的面内C-H弯曲振动出现在范围为1300-1000 cm-1处,1000-750 cm-1为面外摇摆振动[13],Schiff碱苯环上C-H键的面内弯曲振动频率的实验值是1118.48和1078.86 cm-1,对应理论计算值为1116.70和1066.83 cm-1,苯环上C-H键的面外摇摆振动频率的实验值是820.32 cm-1,对应理论计算值818.78 cm-1.芳环骨架的伸缩振动在1650-1450 cm-1之间出现 2~4个吸收峰,由于芳环为一共轭体系,C=C伸缩振动频率位于双键区的低频一端,往往1500 cm-1附近的吸收峰比1600 cm-1强,计算得到的1576.22,1403.85 cm-1峰归属于ph1的伸缩振动,1613.20和1457.80 cm-1为ph2的骨架伸缩振动,1592.48 cm-1为两个苯环骨架振动共同耦合的结果.同时,实验测得的l573.43,1481.31,l454.59和1406.87 cm-1峰与理论计算相符合.芳环的面外弯曲振动在650-900 cm-1,这一区域的吸收峰位置与芳环上取代基性质无关,而与芳环上相连H的个数有关,相连H越多,=C-H 振动频率愈低,计算得到的856.84,814.66,740.34和632.28 cm-1峰归属为苯环的扭曲振动,与实验测量结果878.02,820.32,759.64和600.25 cm-1相一致.
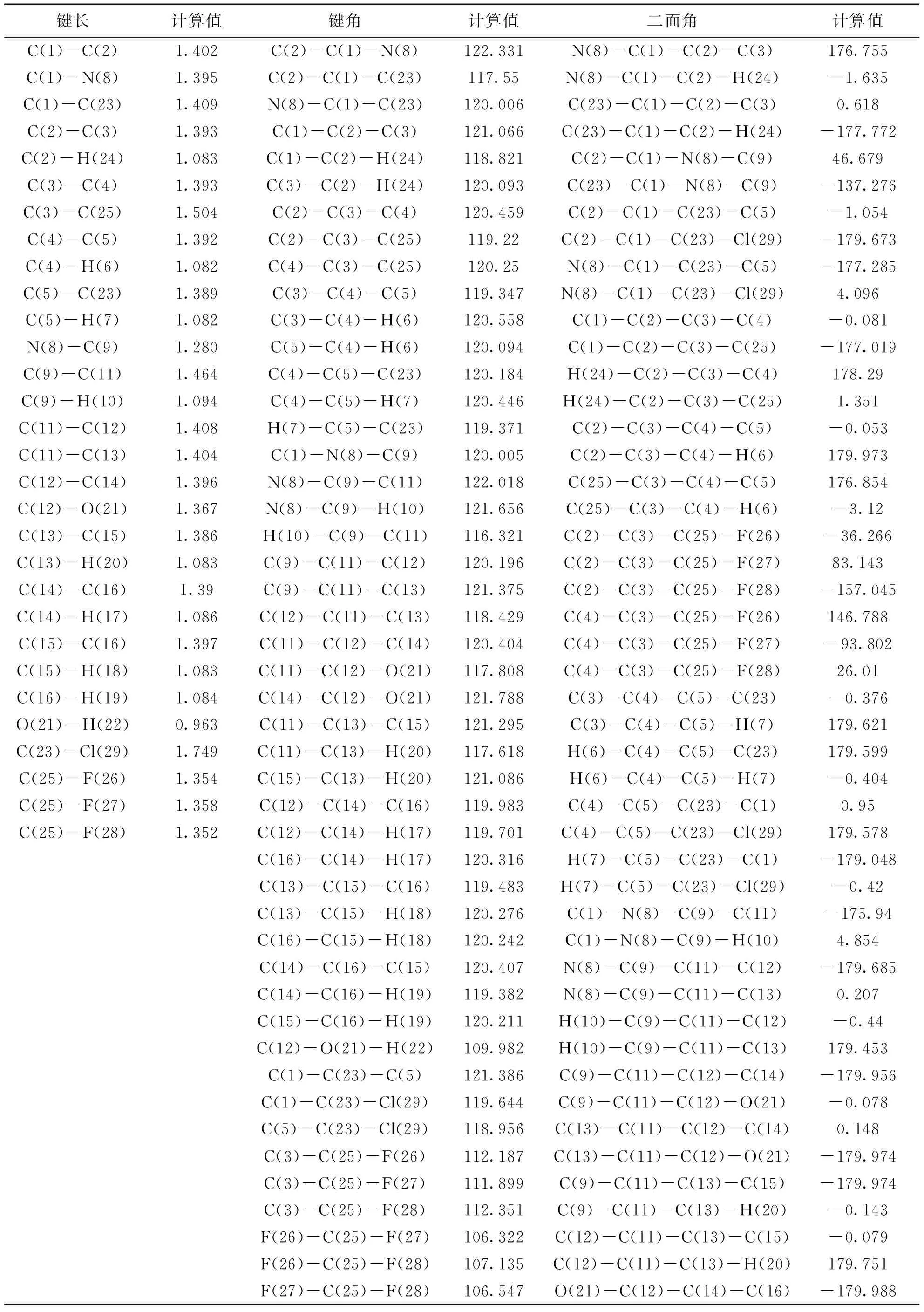
表2 用B3LYP/6-311++G**理论得到的Schiff碱的结构参数
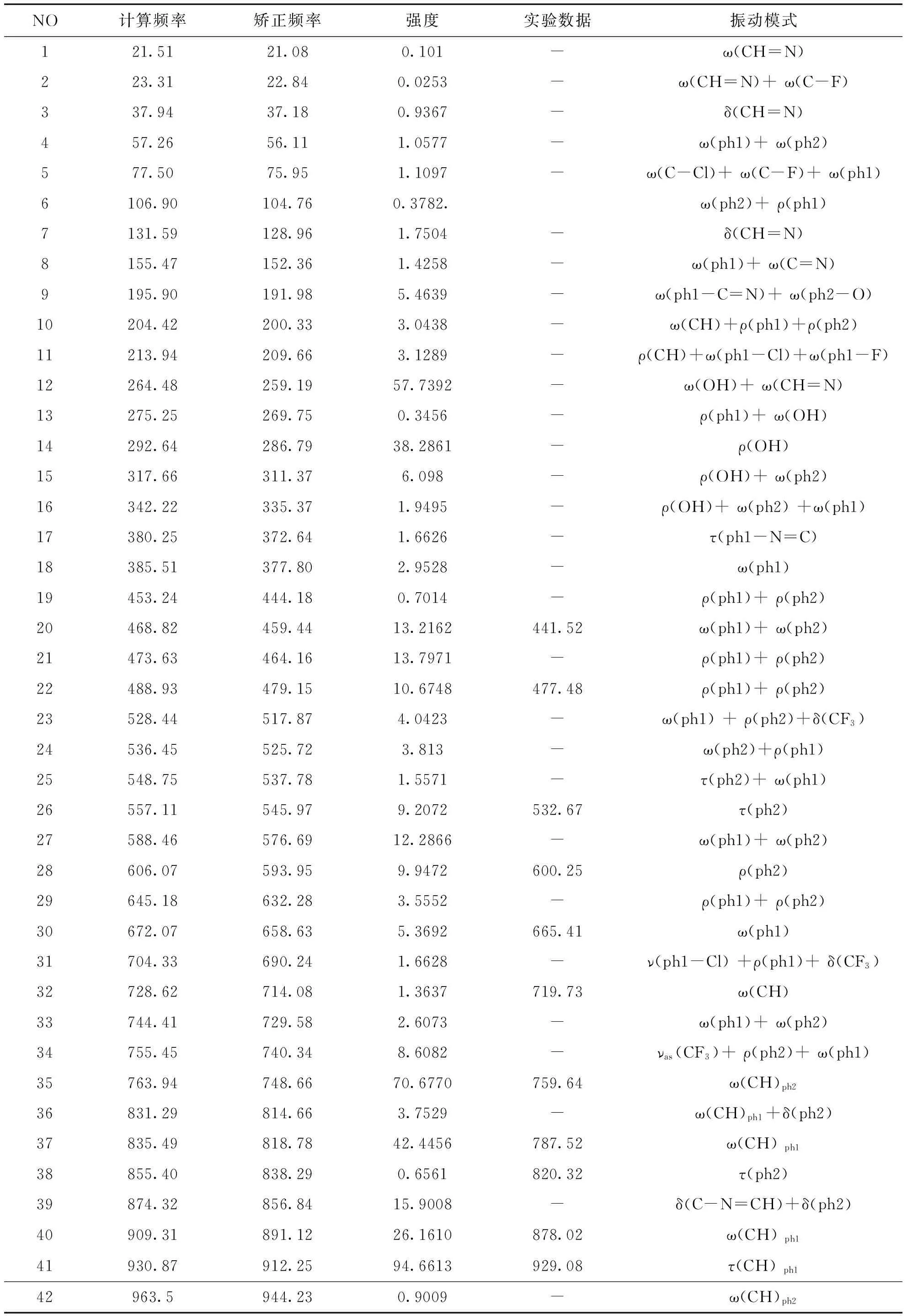
表3 Schiff碱的红外光谱理论数据、实验数据及对应的振动模式
注:ν-伸缩振动;δ-面内弯曲振动;ρ-面内摇摆振动;ω-面外摇摆振动;τ-扭曲振动;s-对称;as-不对称
ph1- 图1a中C1-C2-C3-C4-C5-C23形成的苯环;ph2-图1a中C11-C12-C13-C14-C15-C16形成的苯环
在理论计算中,1648.22 cm-1归属为C9=N8的伸缩振动,该频率也耦合了苯环的振动,实验值为1621.01 cm-1.C-O的伸缩振动在酚和醇中位于1250-1050 cm-1,但受所连基团的影响较大,干扰较多,Schiff碱分子的C-O伸缩振动在实验中是1224.84 cm-1,对应理论计算值1243.45 cm-1,其也耦合了苯环和吡啶环的骨架振动.
Schiff碱分子中的-CF3的振动光谱有显著特征区域.-CF3的对称伸缩在高波数观察区域为1290-1235 cm-1,而不对称伸缩振动在低波数观察区1110-1185 cm-1[14].因此,位于1273.15和1118.48 cm-1的谱带被分配到C-F的对称和不对称伸缩振动,对应理论计算值是1301.57和1116.70 cm-1.-CF3弯曲振动通常发生在区域690-631 cm-1,理论计算为690.24 cm-1,实验值为665.41 cm-1.一般情况下,C-Cl之间的吸收范围是850-550 cm-1[15],因此计算值690.24 cm-1被指定为C-Cl伸缩振动模式.氯代芳烃化合物较强的中等吸收强度在区385-265 cm-1,对于C-Cl面内弯曲振动,对应的频带在311.37 cm-1处,其他的红外光谱理论数据和实验数据列于表3中.
通过对实验频率值和理论计算的结果对比可以发现:从整体上来看,理论计算的频率值与实验结果相吻合.但个别振动频率的计算值和实验值有一定的差别,原因在于理论计算时只考虑到单个分子,没有考虑分子间的作用力,而实验测定分子时,分子间具有相互作用;另外,计算中得到的大多数振动频率都由多个振动模式叠加而成的,也会使理论计算值与实验数据有些不符,由于误差在合理的范围之内,所以对计算结果的分析是合理的.
图3是使用B3LYP/6-311++G**理论计算得到的红外光谱谱图.从图3可以看到,红外光谱的吸收峰分布在波数为3700~3500,3200~3000, l700~800,800~10 cm-1的四个区间内,经分析:在3700~3500,3200~3000区间内的吸收峰主要是与H原子相关的键的伸缩振动,这些振动相对独立,与其他振动形式没有耦合;l700~800,800~10 cm-1两区域内的吸收峰所对应的振动模式比较复杂,基本上每条峰都是由多个振动模式叠加而成的.理论计算谱图与实验谱图相吻合.在计算振动频率方面,B3LYP的计算结果是令人满意的.这说明B3LYP方法在研究相关的Schiff碱体系方面,具有较好的应用性.
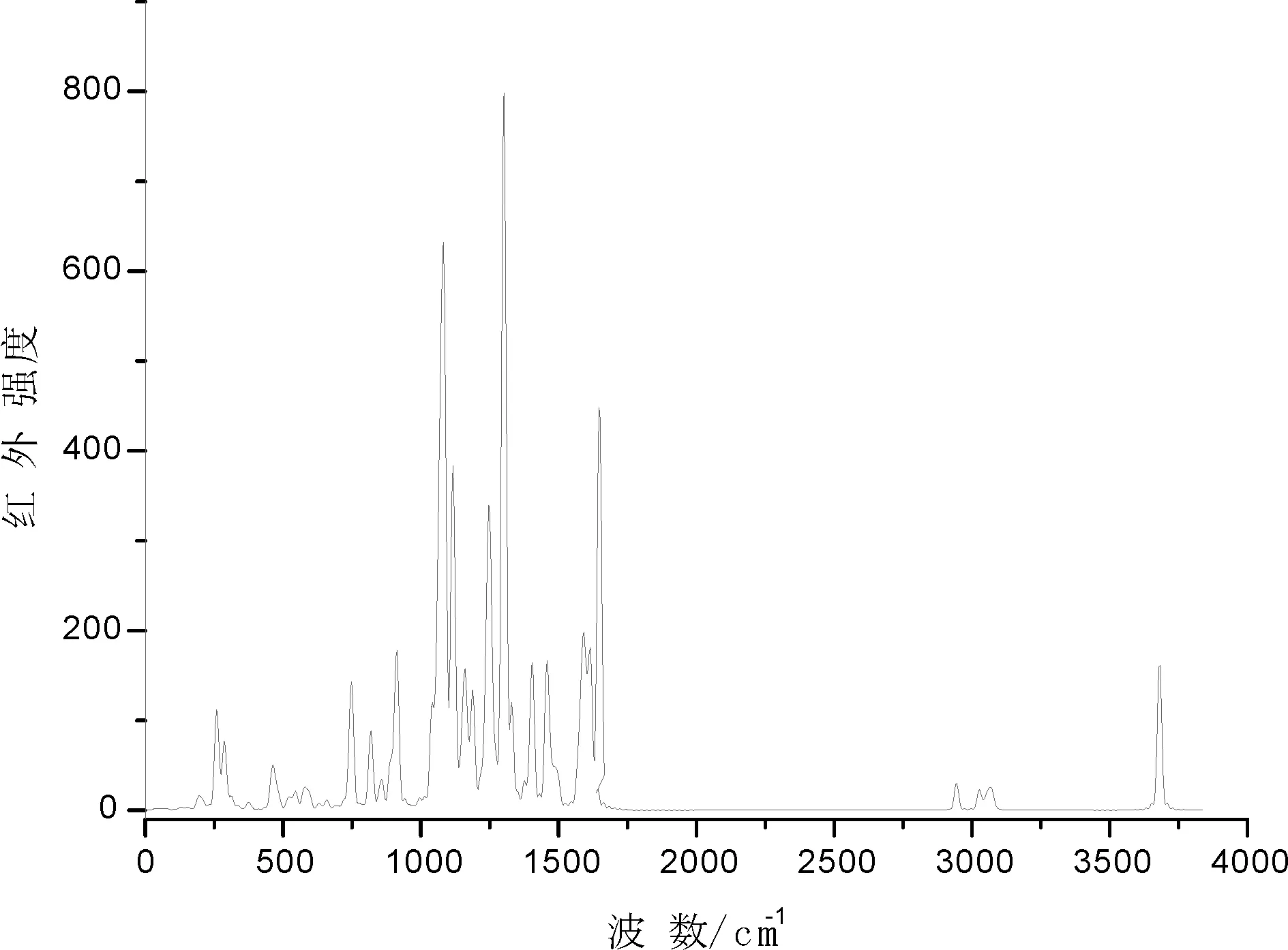
图3 利用B3LYP/6-311++G**理论计算得到的Schiff碱的红外光谱图Fig. 3 Infrared spectrum of Schiff base at B3LYP/6-311++G** level
5 结 论
实验合成了2-氯-5-三氟甲基苯胺水杨醛Schiff碱,测定了红外光谱数据.采用密度泛函理论的B3LYP方法,在6-311++G**基组水平上,对化合物的结构进行优化,得到了其优势构象,并对其稳定构象的键长、键角、二面角进行了分析,Schiff碱分子中两个苯环不在同一平面上,在着绝对值为46.679°的二面角,且分子中存在着较大的共轭体系.结合Gaussian View对红外光谱进行了归属,而且对主要官能团的振动模式进行了详细的讨论.在计算振动频率方面,B3LYP经过校正,理论计算结果和实验数据基本相吻合.目前,量子化学理论计算水平日渐成熟,数据计算精确,为Schiff碱类化合物的进一步研究提供了参考依据.
[1] Keypour H, Rezaeivala M. Synthesis and crystal structure of Mn complexes with novel macrocyclic Schiff base ligand containing piperazine moiety [J].Polyhedron, 2008, 28(17): 3755.
[2] Shridhar M, Arun M, Isloor Shrikrishna I,etal. Synthesis, characterization and antibacterial activity of some new pyrazole based Schiff bases [J].ArabianJournalofChemistry, 2013, 6(3): 335.
[3] Zhong X, An P, Zhang J Q,etal. Synthesis and study on antitumor activities of indolinedione Schiff bases and their complexes [J].ChemicalResearchandApplication, 2012, 24(7): 1046(in Chinese) [钟霞, 安平, 张俊清, 等. 2-吡啶甲醇红外光谱的密度泛函理论研究 [J]. 化学研究与应用, 2012, 24(7): 1046]
[4] Muhammad S, Noor U, Saqib A,etal. Potential bioactive Schiff base compounds: synthesis, characterization, X-ray structures, biological DNA [J].SpectrochimicaActaPartA:MolecularandBiomolecularSpectroscopy, 2013, 116(8): 111.
[5] Antunes J A, Silva L E, Bento R R F,etal. Vibrational spectra and DFT calculations of the vibrational modes of Schiff base C18H17N3O2[J].JournalofMolecularStructure, 2012, 1013: 126.
[6] Li X M, Zhang L B, Zhou L Z,etal. The study of infrared spectra of 2-pyridinemethanol by density functional theory [J].SpectroscopyandSpectralAnalysis, 2012, 32(9): 2358(in Chinese) [李晓明, 张来斌, 周留柱, 等. 2-吡啶甲醇红外光谱的密度泛函理论研究 [J]. 光谱与光谱学分析, 2012, 32(9): 2358]
[7] Zhao B, Wang Y Y, Zhang Y H. Structure and vibrational spectroscopy investigation of 2-(4-chlorophenyliminomethyl)-8-hydroxyquinoline [J].SpectrochimicaActaPartA:MolecularandBiomolecularSpectroscopy, 2011, 81(1): 2511.
[8] Meng D S. Synthesis and characterization of 2-chloro-5- trifluoromethylaniline and its Schiff base [J].JournalofXinyangNormalUniversity:NatruralScienceEdition, 2013, 26(4): 563(in Chinese)[孟德素. 2-氯-5-三氟甲基苯胺及其Schiff碱的合成与表征[J]. 信阳师范学院学报:自然科学版, 2013, 26(4): 563]
[9] Karthick T, Balachandran V, Perumal S,etal. Vibrational (FT-IR and FT-Raman) spectra and quantum chemical studies on the molecular orbital calculations, chemical reactivity and thermodynamic parameters of 2-chloro-5-(trifluoromethyl) aniline [J].SpectrochimicaActaPartA:MolecularandBiomolecularSpectroscopy, 2013, 107(1): 72.
[10] Teimouri A, Chermahini A N, Taban K,etal. Experimental and CIS, TD-DFT, ab initio calculations of visible spectra and the vibrational frequencies of sulfonyl azide-azoic dyes [J].SpectrochimicaActaPartA:MolecularandBiomolecularSpectroscopy, 2009, 72(2): 369.
[11] Dabbagh H A, Teimouri A, Chermahini A N,etal. DFT and ab initio study of structure of dyes derived from 2-hydroxy and 2,4-dihydroxy benzoic acids [J].SpectrochimicaActaPartA:MolecularandBiomolecularSpectroscopy, 2008, 69(2): 449.
[12] Silverstein R M, Webster F X, Kiemle D J.Spectrometricidentificationoforganiccompounds.seventhed. [M]. New York: John Wiley & Sons, 2005.
[13] Krishnakumar V, Prabavathi N. Simulation of IR and raman spectral based on scaled DFT force fields: A case study of 2-amino 4-hydroxy 6-trifluoromethylpyrimidine, with emphasis on band assignmnet [J].SpectrochimicaActaPartA:MolecularandBiomolecularSpectroscopy, 2008, 71(2): 449.
[14] Fernandez L E, Ben Altabef A, Varetti E L. The force constants in the isoelectronic series CF3SO2X (X=F, OH, NH2, CH3): a study based on DFT calculations and experimental data [J].JournalofMolecularStructure, 2002, 612(1) : 1.
[15] Sawant A B, Gill C H, Nirwan R S. Molecular structure and vibrational spectra of 2-[5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl]phenol [J].IndianJournalofPureandAppliedPhysics, 2012, 50(1): 38.
Investigation of structure and vibrational spectroscopy properties of 2-chloro-5-trifluoromethylaniline salicylaldehyde Schiff base
MENG De-Su, LU Jin-Feng
(Department of Chemistry and Chemical Engineering, Heze University, Heze 274015, China)
2-Chloro-5-trifluoromethylaniline salicylaldehyde Schiff base was synthesized with 2-chloro-5-trifluoromethylaniline and salicylaldehyde as raw materials, It is characterized by IR and1HNMR in the experiment. The optimization of the geometries and the calculation of IR of synthetic Schiff bases were performed by Density Functional Theory (DFT) B3LYP method at the 6-311++G**basis set, and the optimized conformations, the frequencies and the corresponding infrared intensities of Schiff base compounds were obtained.The experimental and theoretical calculated spectral data were compared , the vibrational modes were assigned and discussed. it is found that the calculated results were in good agreement with the experimental results.
2-chloro-5-trifluoromethylaniline; Salicylaldehyde; Schiff base; DFT; Vibration spectrum
2014-07-04
山东省教育厅高校科研发展计划项目 (J11LB58);菏泽学院科研基金项目(XY13KJ08)
孟德素(1978—),女,山东菏泽人,硕士,讲师,主要从事有机合成与有机分析的研究.E-mail: desumeng@126.com
103969/j.issn.1000-0364.2015.10.004
O641
A
1000-0364(2015)05-0733-08
猜你喜欢
能源化工(2022年1期)2023-01-14
分子催化(2022年1期)2022-11-02
浙江化工(2022年8期)2022-09-05
化学工业与工程(2022年1期)2022-03-29
能源化工(2021年6期)2021-12-30
纺织检测与标准(2021年3期)2021-12-03
中成药(2019年12期)2020-01-04
中国塑料(2016年2期)2016-06-15
浙江化工(2015年4期)2015-11-28
中国农资(2012年40期)2012-08-15
