
气相中二氧化钒活化甲烷C-H 键的理论研究
2014-07-13 03:39陈晓霞王永成张秀兰
原子与分子物理学报 2014年6期
陈晓霞,王永成,张秀兰
(1. 四川理工学院化学与制药工程学院,自贡643000;2. 西北师范大学化学化工学院,兰州730070)
1 引 言
甲烷具有正四面体的稳定结构(其4 个C –H 键的平均键能为414 kJ·mol-1,第一个C – H键的解离能高达435 kJ·mol-1),甲烷是煤层气和天然气的主要成分,随着石油资源的日益枯竭,储量丰富的天然气资源将成为未来最具有希望的替代能源和化工原料之一[1]. 另外一方面,甲烷可以作为良好的氢气的原材料而吸引了大量的关注[2]. 因此,甲烷高效活化和选择转化成为了催化乃至化学领域的一大难题[3],长期以来一直受到研究工作者的广泛关注和重视,这将会带来极大的经济效益和社会效益[4].
早在1979 年,Allison[5]等人就发现过渡金属阳离子能够活化C-H 键,在接下来的几年里,过渡金属阳离子和小分子烷烃的反应在实验[6]和理论[7]方面都有很多研究. 二十世纪九十年代,SchrÖder 和Schwarz 研究小组已经系统地研究了气相中第一周期过渡金属氧化物离子与甲烷的反应[8-13],随后,Shiota 和Yoshizawa 研究小组的理论计算结果[14,15]与以上实验结论相吻合. 1996年,Fiedler 等[16]首次就过渡金属二氧化物正离子CrO+2 与烷烃小分子的反应进行了实验研究,2006年,王永成研究小组[17]运用量子化学DFT 方法对中间过渡金属氧化物离子活化甲烷这一体系进行了研究,较全面系统地探讨了活化甲烷C-H 键的微观机理. 作为连续报道,本文则探究了前过渡金属氧化物离子在单重态和三重态两个势能面上活化甲烷的三条反应路径:
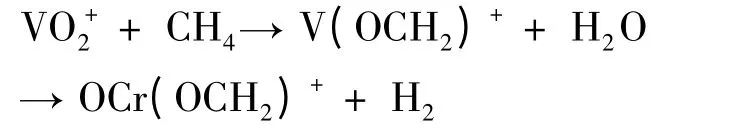
并且对反应过程中发生的势能面交叉现象进行了讨论,深刻揭示活化甲烷C-H 键的微观机理.
2 计算方法
与以往的研究方法[17-20]一样,采用B3LYP方法在6 -311G (2d,p)水平上,对单重态和三重态两个反应势能面上的反应物、中间体、过渡态和产物的构型进行了全参数优化,并通过频率分析,证实了各反应物、中间体和产物的能量是局部极小,各过渡态构型有唯一虚频. 并且在B3LYP/6 -311 + +G (3df,3pd)水平上计算了各驻点的总能量. 两个势能面上的反应物、中间体、过渡态及产物的主要结构参数见图1 和图2.
3 结果与讨论
3.1 反应物优化结果
根据文献[21]提供的信息,反应物VO+2存在两种不同自旋多重度的基态(图1):单重态和三重态. 且利用B3LYP 方法在6 -311G (2d,p)水平上优化得出了这两种自旋多重度不同的反应物的结构,其中单重态属于C2V点群,对称性较高,两个V-O 键长相等为0.1546nm,O -V -O键角为106.2°,能量较低;三重态属于CS点群,对称性较低,键长不等,分别为0.1763 nm 和0.1529nm,键角略大,为110.0°,能量较单重态高143.4kJ·mol-1. 单重态的VO+2为其更稳定的基态.
3.2 反应机理
根据大量计算得出VO+2活化甲烷可以得到两种不同的产物且有三条不同的反应通道. 由于反应物VO+2存在两种自旋多重度的基态,所以反应可以发生在两个不同的势能面上. 在第一条反应路径(图3a)中:两个不同的反应势能面上的反应路径很相似. 首先,反应物VO+2(C2v,1A1)/(A″)和CH4形成反应物复合物1IM1/3IM1,接着1IM1/3IM1中与碳原子相连的一个H 原子向其中一个O 原子(1 号O 原子)迁移而使C-H 键断裂并形成O-H 键,经过能垒为126.4 kJ·mol-1/66.9 kJ · mol-1的 过 渡 态1TS1/3TS1形 成 了 中 间体1IM2/3IM2. 然后,随着O-V-O 键角逐渐增大,V 原子向另一个O 原子(2 号O 原子)不断靠近而成键,经过过渡态1TS2/3TS2而形成中间体1IM3/3IM3. 随着反应的进行,与碳原子相连的另一个H 原子向1 号O 原子迁移而使第二个C-H 键断裂,经过能垒为190.7 kJ·mol-1/173.1 kJ·mol-1的过渡态1TS3/3TS3,V-O1键逐渐地被拉长而断裂形成产物复合物1IM4/3IM4,最终生成产物P1 [V(OCH2)++H2O].
相比于第一条反应路径,第二条反应路径(图3b)与其产物相同,但具体的反应历程却不同. 生成1IM1/3IM1后,反应首先经历了1TS4/3TS4这一过渡态,使得与碳原子相连的一个H 原子向V原子方向迁移而使C-H 键断裂,同时C 原子与2号O 原子逐渐形成C -O 键而生成1IM5/3IM5. 接着,这个H 原子继续向1 号O 原子迁移并形成O-H 键而生成1IM6/3IM6. 当经过C -O -V 键角弯曲振动的过渡态1TS6/3TS6,C -O -V 键角发生较大弯曲变为几乎90°而形成中间体1IM3/3IM3. 不过在单重态势能面上,1IM6经过1TS6直接生成的是1IM7,它和1IM3是一对对映异构体. 后面的历程和第一条路径是相同的,经过TS3到达IM4再生成产物P1. 在这条反应路径中虽然IM5容易生成IM6,之后反应经历的过渡态能垒也不是很高,但是IM1生成IM5经过的过渡态1TS4/3TS4的能垒却高达241.5 kJ·mol-1/268.8 kJ·mol-1,所以这条反应路径也不易发生.
第三条路径(图3c)中反应开始的方式与第二条路径的开始阶段相同. 反应生成1IM5/3IM5后,经历了1TS8/3TS7这一过渡态,与碳原子相连的另一个H 原子向2 号O 原子迁移而使第二个CH 键断裂并形成了O-H 键,生成了1IM8/3IM7. 这以后的反应路径就较为平坦了,这个被解离出来的H 原子继续向V 原子方向迁移而形成了1IM9/3IM8.接着分别处于V 原子两边的两个H 原子不断远离V 原子并成键,最终生成了产物P2 [OV(OCH2)++H2]. 第三条反应路径与前两条相比很明显是一条放热的(-67.8 kJ·mol-1)反应路径,但反应经历的过渡态TS4的能垒较高,因此整条通道是一条热力学较易发生而动力学不易发生的反应通道.
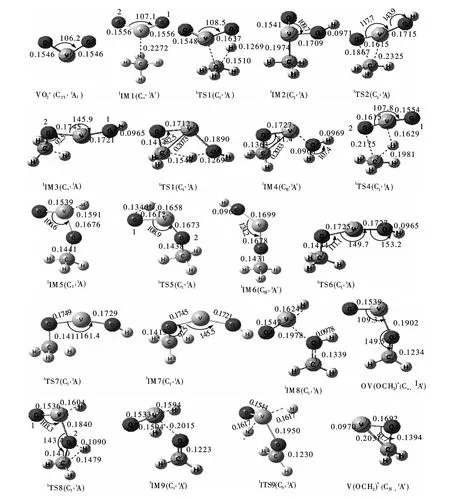
图1 B3LYP/6 -311G (2d,p)水平上单重态势能面上各驻点的几何构型(键长单位:nm,键角单位:°)Fig.1 Geometries of the critical points on the singlet PES at the B3LYP/6 -311G (2d,p)level (Bond lengths are given in nm and angles in degrees)
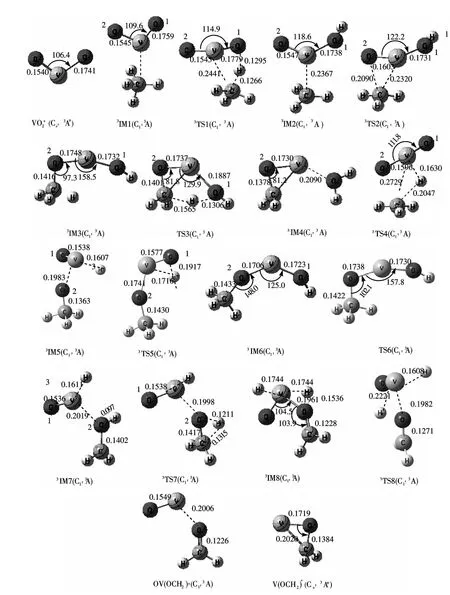
图2 B3LYP/6 -311G (2d,p)水平上三重态势能面上各驻点的几何构型(键长单位:nm,键角单位:°)Fig.2 Geometries of the critical points on the triplet PES at the B3LYP/6 -311G (2d,p)level (Bond lengths are given in nm and angles in degrees)
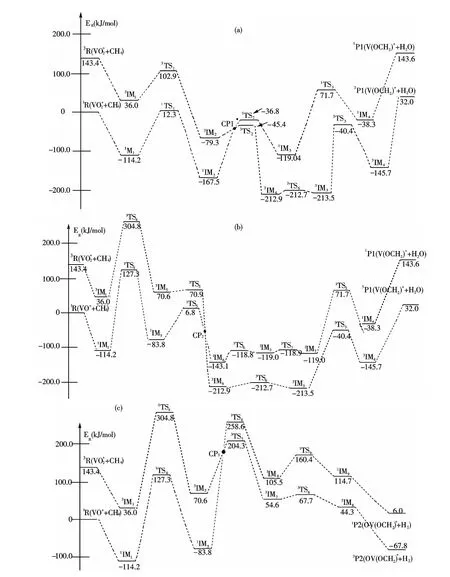
图3 经B3LYP/6 -311 + +G (3df,3pd)能量校正后的和CH4 在两个势能面上的三条反应路径示意图Fig.3 Diagram of the path channels for the reaction + CH4 on both the singlet and triplet PESs at the B3LYP/6 -311 + +G (3df,3pd)level
3.3 势能面交叉现象
运用B3LYP/6 -311 + +G (3df,3pd)方法计算可知,三重态(Cs,3A'')的能量比单重态(C2v,1A1)的能量高,在路径一中3IM2和1IM2的能量分别为-79.3 kJ·mol-1和-167.5 kJ·mol-1,3IM2的能量高于1IM2的,但是3IM3的能量为- 215.3 kJ·mol-1却低于1IM3的,为-119.0 kJ·mol-1. 根 据Hammond[22]假 设,推断出在反应过程中发生了势能面交叉现象,属于Hammond 假设的第三种情况,即交叉现象出现在过渡态之前,且是一个典型的两态反应[22]. 大致的势能面交叉位置在IM2→TS2 过程中CP1 附近.运用同样的方法,找到路径二中发生势能面交叉现象大致的位置在TS5 →IM6 过程中CP2 附近,属于Hammond 假设的第二种情况,即交叉现象出现在过渡态之后. 还有路径三中发生势能面交叉现象大致的位置在IM5 →TS7 过程中CP3 附近,属于Hammond 假设的第三种情况,即交叉现象出现在过渡态之前. 总观三条反应路径,当VO+2在单重态势能面上插入到CH4的C -H 键生成稳定的中间体(IM2、IM5)后,第一个C -H 键被活化了,随着反应体系通过“系间窜越”转到能量较低的三重态势能面上进行,第二个C -H 键的活化发生在更为平坦的三重态势能面上,由于CP1,CP2 和CP3 都出现在反应较前面的阶段,所以势能面交叉附近的自旋翻转成为了控制整个反应速率的因素之一.
4 结 论
采用密度泛函B3LYP/6 -311 + + G (3df,3pd)//6 - 311G (2d,p)研究了气相中(1A1/3A')活化甲烷生成P1 [V (OCH2)++H2O]和P2 [(OCH2)++H2]的反应. 探索了三条反应路径,其中生成P1 产物有两条反应通道. 相比而言,生成P2 产物,即第三条路径为较有利反应通道. 其结果与前过渡金属离子CrO+2+CH4体系[17]的反应机理极其相似,且反应过程中都发生了势能面交叉现象,甲烷的C -H 键的活化过程同样涉及自旋多重度的改变,且是一个典型的TSR 反应. 研究结果既可以使我们深刻理解该反应的反应机理,为以后的实验研究提供有用的理论依据,同时也可以为VO+2与其他饱和烃和不饱和烃反应的实验研究提供帮助.
[1] Li W Z. Recent development in the catalytic conversion of natural gas[J]. Chemical Engineering of Oil and Ges,1998,27(1):1(in Chinese)[李文钊. 天然气催化转化新进展[J]. 石油与天然气化工,1998,27(1):1]
[2] Forster P,Ramaswamy V,Artaxo P,et al. Changes in atmospheric constituents and in radiative forcing[J].Climate change 2007:The physical science basis. Contribution of working group I to the forth assessment report of the intergovernmental panel on climate change,129.
[3] Leny Y,Hisao Y. Photocatalytic conversion of methane[J]. Chem. Soc. Rev.,2008,37:1592.
[4] Crabtree R H. Aspects of methane chemistry [J].Chem. Rev.,1995,95:987.
[5] Allison J,Freas R B,Ridged P. Cleavage of alkanes by transition metal ions in the gas phase[J]. J. Am.Chem. Soc.,197,101(5):1332.
[6] Squires R R. Gas - phase transition - metal negativeion ion chemistry [J]. Chem. Rev.,1987,87(3):623.
[7] Hendrickx M,Ceulemans M,Gong K,et al. Theoretical study on the stability of low - spin hydridomethyl complexes of the first - row transition metal cations[J]. J. Phys. Chem. A,1997,101(13):2465.
[8] Schröder D,Schwarz H. FeO+activates methane[J].Angew. Chem. Int. Ed. Engl.,1990,29:1433.
[9] Schröder D,Fiedler A,Hrusók J,et al. Experimental and theoretical studies toward a characterization of conceivable intermediates involved in the gas -phase oxidation of methane by bare FeO+. Generation of four distinguishable[Fe,C,H4,O]+isomers[J]. J.Am. Chem. Soc.,1992,114:1215.
[10] Ryan M F,Fideler A,SchrÖder D,et al. Stoichiometric gas-phase oxidation reactions of CoO+with molecular hydrogen,methane,and small alkanes[J].Organometallics,1994,13:4072.
[11] Ryan M F,Fideler A,Schröder D,et al. Radical -like behavior of manganese oxide cation in its gas -phase reactions with dihydrogen and alkanes[J]. J.Am. Chem. Soc.,1995,117:2033.
[12] Schwarz J,Wesendrup R,SchrÖder D,et al. Mass spectrometric study of[Fe,C3,H6,O]+ isomers relevant in the gas -phase oxidation of hydrocarbons by“Bare”FeO+[J]. Chem. Ber.,1996,129:1463.
[13] Schröder D,Schwarz H Angew. C-H and C-C bond activation by bare transition - metal oxide cations in the gas phase[J]. Chem. Int. Ed. Engl.,1995,34:1973.
[14] Yoshizawa K,Shiota Y,Yamabe T. Methane-methanol conversion by MnO+,FeO+,and CoO+:A theoretical study of catalytic selectivity[J]. J. Am. Chem.Soc.,1998,120:564.
[15] Shiota Y,Yoshizawa K. Methane-to-methanol conversion by first - row transition - metal oxide ions:ScO+,TiO+,VO+,CrO+,MnO+,FeO+,CoO+,NiO+,and CuO+[J]. J. Am. Chem. Soc.,2000,122:12317.
[16] Fiedler A,Kretzschmar I,SchrÖder D,et al. Chromium dioxide cation OCrO+in the gas phase:Structure,electronic states,and the reactivity with hydrogen and hydrocarbons[J]. J. Am. Chem. Soc.,1996,118:9941.
[17] Chen X X,Wang Y C,Geng Z Y,et al. Theoretical study of gasphase activation of C-H bond of methane by[J]. Acta Chim. Sin.,2006,64:637(in Chinese)[陈晓霞,王永成,耿志远,等. 气相中活化甲烷C - H 键的理论研究[J]. 化学学报,2006,64(7):636]
[18] Chen X X,Feng X S,Gao L G,et al. A theoretical study of C - C bond activation of C2H4in gas phase[J]. Journal of Molecular Structure:THEOCHEM,2009,906:31.
[19] Chen X X. Theoretical study on the mechanism of the+ H2reaction in gas phase[J]. Chinese J.Struct. Chem.,2012,31(2):161.
[20] Chen X X. A theoretical study of H-H σ bond activation catalyzed byin gas phase[J]. International Journal of Quantum Chemistry,2012,112:359.
[20] Gracia A,Sambrano J R,Safont V S,et al. Theoretical study on the molecular mechanism for the reaction ofwith C2H4[J]. J. Phys. Chem. A,2003,107:3107.
[22] Schröder D,Shaik S,Schwarz H. Two-state reactivity as a new concept in organometallic chemistry[J].Acc. Chem. Res.,2000,33:139.
猜你喜欢
高中数理化(2022年16期)2022-09-14
北京航空航天大学学报(2022年5期)2022-06-06
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22
电脑知识与技术(2018年3期)2018-03-21
中学化学(2017年5期)2017-07-07
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
中学化学(2016年4期)2016-05-30
中学化学(2014年1期)2014-04-23
