
VEGFR-2酪氨酸激酶抑制剂研究进展
2014-03-14 09:48叶炎钊施志浩陆涛
药学进展 2014年1期
叶炎钊, 施志浩, 陆涛
(中国药科大学理学院, 江苏 南京211198)
·综述与专论· REVIEW AND MONOGRAPH
VEGFR-2酪氨酸激酶抑制剂研究进展
叶炎钊, 施志浩, 陆涛*
(中国药科大学理学院, 江苏 南京211198)

血管内皮生长因子受体是一种可诱导血管生成的受体型酪氨酸激酶, 临床研究发现该激酶在肿瘤组织中过度表达。以抑制血管内皮生长因子受体为基础的抗血管生成疗法已成为癌症治疗的新策略, 小分子血管内皮生长因子受体-2抑制剂的设计和合成获得了广泛关注。对近5年内血管内皮生长因子受体-2抑制剂的研究进展进行了综述。
抗肿瘤;血管生成抑制剂;VEGFR-2;酪氨酸激酶抑制剂
研究显示:VEGFR-2活性腔主要由疏水腔组成, 与配体结合相关的主要有4个子区域:Ⅰ区为与绞链区毗邻的1个扁平的疏水腔, 由Val846、Ala864、Val897、Val914、Phe916、Leu1033的疏水性侧链组成(陈军, 化学学报, 2007年)。其中包含2个关键的氢键作用位点:Glu915的C=O作为氢键受体, Cys917 的NH作为氢键供
血管内皮生长因子(vascular endothelial growth factor, VEGF)是目前发现的作用最强、特异性最高的促血管生成因子。VEGF在脉管生成、血管形成和血管迁移过程中起重要调节作用, 并在多种恶性肿瘤中过度表达, 与肿瘤的生长、转移、预后关系密切。VEGF家族包括VEGF-A、VEGF-B、VEGF-C、VEGF-D、VEGF-E及胎盘生长因子(PIGF)。
VEGFR是一类酪氨酸激酶跨膜糖蛋白, 它由7个Ig结构域组成的胞外区、一个跨膜结构区和胞质内酪氨酸激酶结构区组成。VEGFR主要包括VEGFR-1(Flt-1)、VEGFR-2(KDR/Flk-1)、VEGFR-3(Flt-4) 这3种受体,体。Ⅱ区是由Ile886、Leu887、Ile890、Val896、Val897、Leu1017、Ile1042的疏水性侧链组成的一个较大的疏水腔。Ⅰ区和Ⅱ区之间有1个由Lys866、Glu883的侧链和Asp1044骨架上的羰基氧形成的重要的极性区域。其中Asp1044侧链上的NH可以作为1个关键的氢键供体位点, Glu883的C=O作为氢键受体。Ⅲ区是1个小的疏水性区域, 由Leu838和Phe916的侧链组成。Ⅳ区是由疏水的Leu1033、Cys1043和极性的Asn921、Arg1030、Asn1031组成的一个极性区域。小分子化合物通过与VEGFR-2蛋白活性位点结合发挥抑制作用。
在过去数年中, 已报道了大量的VEGFR-2抑制剂, 部分抑制剂已进入临床研究阶段[2-4], 本文根据作用机制及分子骨架的不同, 对近5年来报道的VEGFR-2小分子抑制剂的研究进展进行综述。
1 可逆抑制剂
可逆抑制剂的作用机制是通过与ATP 竞争性结合VEGFR-2在胞外的配体结合位点, 阻断分子内酪氨酸的自身磷酸化, 干扰酪氨酸激酶活化, 抑制VEGFR 激活, 从而达到破坏细胞周期进程、加速细胞凋亡、抑制血管生成及抑制肿瘤浸润和转移的作用。此类抑制剂是当前研究的热点, 根据分子骨架不同, 分为以下几类。
1.1 二芳基脲类
二芳基脲类化合物是由拜耳公司通过高通量筛选技术发现的一类Raf激酶抑制剂, 后来发现此类药物对VEGFR也有较强的抑制作用。其中, sorafnib(1)于2005年获美国FDA批准用于治疗晚期肾细胞癌, 并于2007年获准用于治疗肝细胞癌(Wilhelm S, Nat Rev Drug Discov, 2006年);2012年9月, 美国FDA批准口服药物regorafenib(2)用于治疗转移性结直肠癌(Wilhelm S, Mol Cancer Ther, 2007年);目前, 由日本卫材公司开发的lenvatinib(3)已进入Ⅱ期临床(Matsui J, Clin Cancer Res, 2008年), tivozanib(4)已进入Ⅲ期临床阶段, 它们均可选择性抑制VEGF介导的VEGFR磷酸化过程, 阻断VEGF诱导的MAPK通路的激活, 从而抑制内皮细胞增殖(Nakamura K, Cancer Res, 2006年);化合物ABT-869(linifanib, 5)是一种多靶点酪氨酸激酶抑制剂, 对所有VEGFR和PDGFR家族的成员均有很强的阻断活性, 对VEGF刺激的内皮细胞增殖的阻断作用很强(IC50=0.2 nmol·L-1), 对无关受体酪氨酸激酶、可溶性酪氨酸激酶和丝/苏氨酸激酶的阻断活性较差(IC50>2 μmol·L-1)。ABT-869可特异性地通过阻断新生血管生成切断肿瘤的血液供应, 从而抑制肿瘤细胞的生长和转移。该化合物现已进入Ⅲ期临床研究阶段[5]。
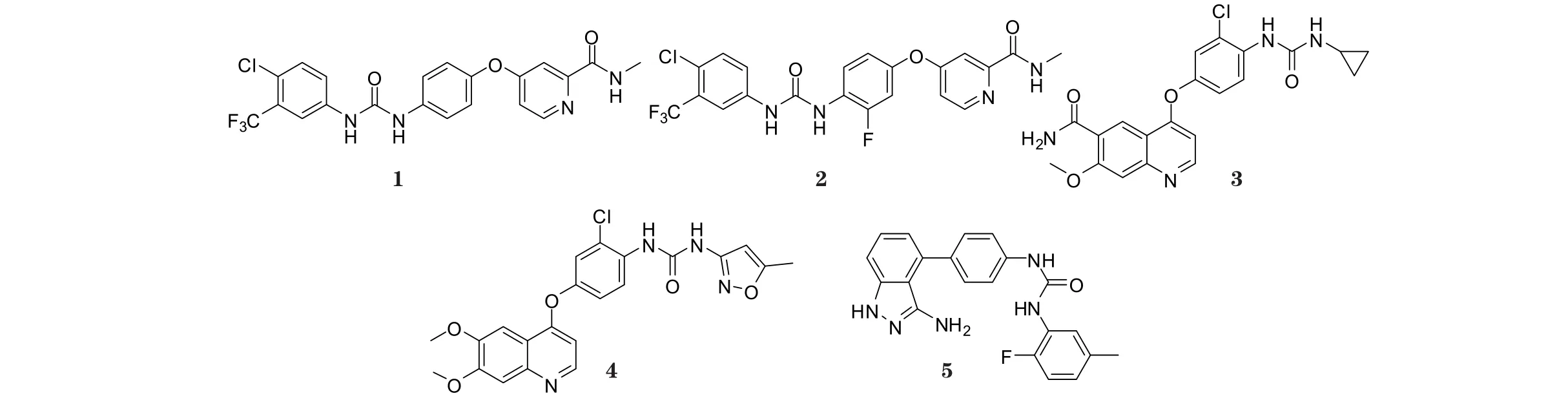
雅培公司(Abbot)的研究人员也报道了一系列与ABT-869结构类似的N, N ′-二芳基脲类抑制剂, 这些化合物对VEGFR和PDGFR家族均具有较高活性, 其中化合物6~8对VEGFR-2的IC50分别为3、45和11 nmol·L-1(Heyman H R, Bioorg Med Chem Lett, 2007年;Dai Y, Med Chem, 2005年;Ji Z, J Med Chem, 2008年), 这些化合物可通过其脲结构延伸到酶的ATP结合位点的疏水口袋, 从而抑制酶的活性。
化合物9是由武田制药开发的VEGFR-2激酶抑制剂,对VEGFR-2、VEGFR-1、PDGFRα和PDGFRβ均有一定抑制作用, IC50分别为6.2、15、35和96 nmol·L-1。研究显示, 化合物9中脲基团通过氢键占据疏水口袋;吡咯并嘧啶母核1位N和Cys919在铰链区以氢键相连接;脲基团和蛋白质通过2个氢键结合, 脲的NH连接Glu885,羰基连接Asp1046, 3-三氟甲基苯基部分占据着一个由Phe1047构象重排产生的疏水性口袋。该化合物可抑制HUVEC细胞的增殖, 其体外IC50为4.4 nmol·L-1[6]。
化合物10具有对VEGFR-2和FGFR-1的双重抑制作用(体外IC50分别为9.3和14 nmol·L-1)。该化合物在末端的苯环上有一个哌嗪, 其可与VEGFR-2上的Ile1025、His1026以及FGF-1上的Ile620、His621相互作用, 从而抑制VEGF和FGF与其受体结合, 阻止HUVEC的增生。FGF-FGFR信号通路不仅促进肿瘤血管增生, 且有利于肿瘤的生长和存活, 化合物10可抑制该受体的酪氨酸激酶活性, 从而有效抑制肿瘤的生长[7]。
化合物11对VEGFR-2和KDR细胞均具有抑制活性,体外IC50分别为3和0.7 nmol·L-1(Frey R R, J Med Chem, 2008年)。本品具有良好的药动学特性, 其由化合物12改造而来。分子模拟显示, 化合物11的N, N’-二芳基脲占据了VEGFR-2的疏水口袋。研究显示, 化合物11的7-氨基吡唑并嘧啶母核能与酶ATP口袋的铰链区形成氢键;此外, 氨基和Glu915羰基之间, 以及吡唑并嘧啶的N1和Cys917的NH之间均存在氢键的相互作用(见图1)。

图1 化合物11与VEGFR-2的结合模型Figure 1 The model of compound 11 binding with VEGFR-2
化合物13的吲唑环的NH和N分别和Glu917和Cys919形成氢键, 与Leu840、Val848、Ala866、Lys868、Glu917、Phe918和Gly922发生疏水作用, 三唑的2位N和Lys868形成氢键, 脲的NH和Lys868分别形成2个氢键,苯环部分与Ile888、Ile892、Val898、Val899、Leu1019、His1026、Ile1044、Cys1045和Phe1047发生疏水作用。化合物13对VEGFR-2和HUVEC均有抑制活性, 体外IC50分别为0.56和0.3 μmol·L-1。构效关系研究显示, 在三唑环上引入吲唑环, 所得化合物活性高于引入其他芳环, 且吲唑环的立体构型对化合物的活性影响较大, NH和三唑环的N=N在同侧时, 化合物对VEGFR-2的抑制活性最强[8]。化合物14对VEGFR-2具有抑制作用(体外IC50为28 nmol·L-1), 研究发现, 在其脲结构中NH上连接异唑基团等芳杂环时, 可增强化合物的活性;侧链上引入吗啉基团可增加化合物的水溶性, 从而提高其生物利用度[9]。

1.2 吲哚酮类
该类化合物有一个六元芳环骈五元环的稠环母核(以氧代吲哚环为代表), 其1位的NH和VEGFR-2铰链区Glu917的C=O形成分子间氢键, 吲哚酮环的2位羰基氧与Cys919的NH形成一个氢键。在稠环母核中, 五元环以双键桥接吡咯及其衍生物或各种其他取代基团时, 化合物活性较好。吲哚酮类衍生物对VEGFR-2具有抑制作用,以化合物15为例, 其1位NH和KDR铰链区Glu917形成分子间氢键, 吡咯环上的NH和Cys919形成氢键, 5位或6位上的疏水性取代基与由Val899、Phe1047、Leu889、Val914的疏水性侧链以及Lys868的疏水侧链部分组成的一个空腔发生疏水作用。
舒尼替尼(sunitinib, 商品名:Sutent, 代号:SU11248, 15)为辉瑞公司开发的吲哚酮类衍生物, 对VEGFR-2有特异性抑制作用(体外IC50为2~17 nmol·L-1), 本品于2006年1月获美国FDA批准上市, 用于治疗胃肠道基质肿瘤和转移性肾细胞癌(Motzer R J, JAMA, 2006年)。Orantinib(SU6668, TSU-68, 16)是由Sugen公司开发的口服小分子多靶点酪氨酸激酶抑制剂, 其结构类似于舒尼替尼, 不同之处在于化合物的吡咯部分接有羧基, 对VEGFR-2、PDGFRβ和FGFR-1酪氨酸激酶均具有抑制作用, 其IC50达亚微摩尔水平(Sun L, J Med Chem, 1999年)。目前正在进行有关orantinib的Ⅰ/Ⅱ期临床试验, 初步结果显示, 其对实体瘤尤其是肝癌有一定疗效且安全性较高,和其他抗肿瘤药物合用治疗乳腺癌也获得了阳性结果。
Nintedanib(BIBF1120, 17)是由勃林格殷格翰公司开发的一种6-甲氧基羰基取代的吲哚酮衍生物, 其C6位的羰基氧能与Lys868形成额外的氢键。该化合物是一种能作用于VEGFR、PDGFR和FGFR的三重血管激酶抑制剂, 其对VEGFR-1、VEGFR-2、VEGFR-3、FGFR-1和PDGFRα的IC50分别为104、5、5、38和18 nmol·L-1。Nintedanib对VEGF敏感的膀胱内皮细胞和平滑肌内皮细胞均具有良好的细胞增殖抑制活性, 本品口服有效且在体内肿瘤模型中表现出一定的疗效和良好的耐受性。目前正在进行关于nintedanib的Ⅲ期临床研究, 考察其治疗非小细胞肺癌(NSCLC)、卵巢癌及其他恶性实体瘤的疗效[10]。

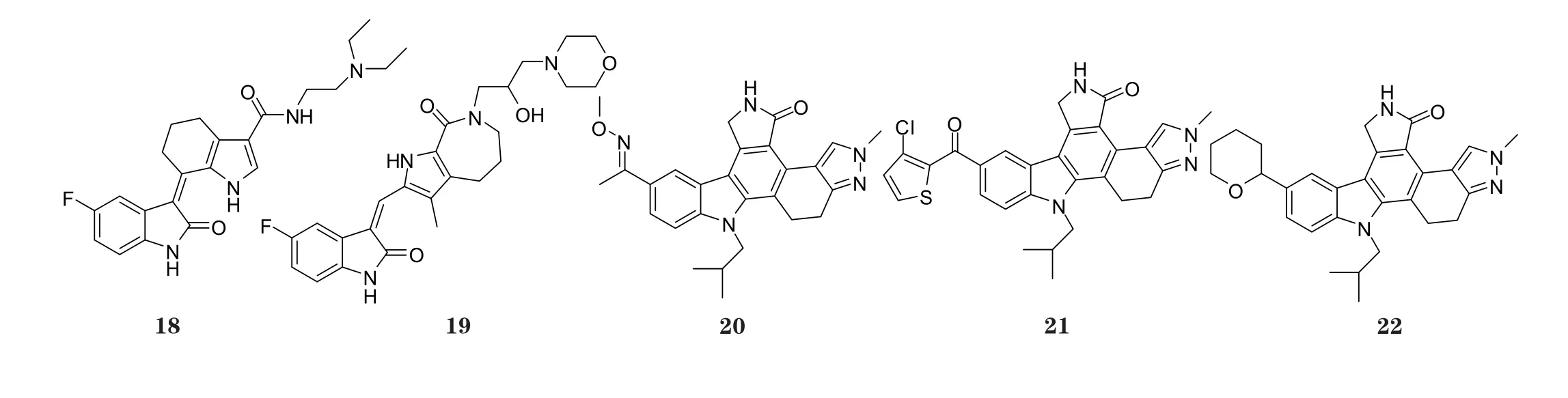
化 合 物18对VEGFR-2、PDGFR、c-Kit、RET和FLT3具有抑制作用(体外IC50为5~68 nmol·L-1), 在动物模型中进行的研究显示, 本品能对内皮细胞的增殖和趋化、角膜的血管生成和肿瘤的生长产生抑制作用[11]。化合物19对结肠癌HT-29细胞具有良好的增殖抑制作用, 其对VEGFR-2和HUVEC均具有抑制活性(体外IC50分别为27和26 nmol·L-1)[12]。化合物20和21为Tie-2和VEGFR-2的双重抑制剂, 其吲哚环作为疏水区与Phe1047通过π-π键发生相互作用, 从而发挥激酶抑制活性, 这2个化合物对VEGFR-2的IC50均为7 nmol·L-1(Dandu R, Bioorg Med Chem Lett, 2008年;Underiner T L, Bioorg Med Chem Lett, 2008年)。化合物22亦为Tie-2和VEGFR-2的双重抑制剂, 作用机制与化合物20和21相同, 其对VEGFR-2的IC50为21 nmol·L-1[13]。
1.3 嘧啶衍生物
1.3.1 喹唑啉类 研究发现, 喹唑啉类抑制剂苯胺部分可进入激酶活性口袋底部, 能够与受体残基非极性侧链产生较强的范德华力和疏水作用。喹唑啉环上1位N原子能够和Met769上的NH形成稳定氢键, 3位N原子则和周围的一个水分子形成氢键。同时, 芳环上的取代基也能和活性口袋外部的部分残基形成一定的范德华力和疏水作用。
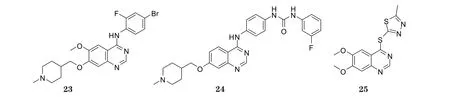
Vandetanib(商品名:Zactima, 23)是由AstraZeneca公司开发的一种口服的小分子多靶点酪氨酸激酶抑制剂,对EGFR、VEGFR-2和胶质细胞源性神经营养因子酪氨酸激酶受体(RET)均有拮抗作用。本品是首个获FDA批准用于治疗晚期转移性甲状腺癌的药物[14]。目前, 我国正在进行vandetanib治疗晚期乳腺癌、NSCLC与晚期多发性骨髓瘤的Ⅲ期临床试验[15]。在vandetanib结构基础上衍生出一系列4-氨基喹唑啉类VEGFR抑制剂, 其中, 化合物24的结构中引入了脲基团, 该化合物对VEGFR-2和VEGFR-3均有一定的抑制作用(体外IC50分别为5.5和9.6 nmol·L-1)[16]。SKLB1002(25)是一种结构较为简单的喹唑啉类选择性VEGFR-2抑制剂(体外IC50为32 nmol·L-1), 其对其他类型的激酶抑制活性较低。本品具有显著的抗血管生成活性, 在10 μmol·L-1的浓度下对脐静脉内皮细胞血管形成的抑制率达98%[17]。
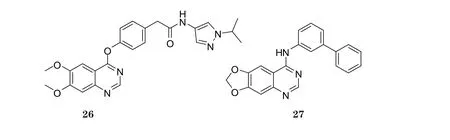
阿斯利康最近报道, 其研发的AZD2932(26)对VEGFR-2和PDGFR有显著的抑制活性, IC50分别为8和4 nmol·L-1。构效关系研究显示, 在酰氨基上连接吡唑环有利于化合物脂溶性的提高, 吡唑环的NH烷基化后可使化合物活性提高[18]。化合物27对VEGFR-2具有较强的抑制活性(体外IC50为0.8 nmol·L-1), 该化合物的喹唑啉环能和Cys917形成氢键, 联苯基团可与Phe1045和Gly1046形成范德华力[19]。
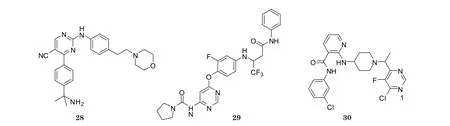
1.3.2 非稠合的嘧啶类 孤立的嘧啶环出现在许多VEGFR-2抑制剂中。苯乙基吗啉衍生物28对VEGFR-2具有一定的抑制作用(体外IC50为27 nmol·L-1), 并对体内新生血管的生成有抑制活性(Hughes T V, Bioorg Med Chem Lett, 2007年)。化合物29能降低VEGF依赖的静脉内皮细胞的增殖(IC50为40 nmol·L-1)。在体外血管生成实验中, 化合物29显示出抑制毛细血管生长的作用[20]。BRN-250(30)为烟碱类酪氨酸激酶抑制剂, 对VEGFR-2和HUVEC具有抑制作用。BRN-250酰胺键上的NH能与Glu883形成氢键, 嘧啶环上的1位N原子与Cys917形成氢键, 吡啶环连接的NH与Asp1044形成氢键[21]。
1.4 吡啶衍生物
1.4.1 喹啉类 喹啉类VEGFR抑制剂是在喹唑啉的基础上设计出来的。在对喹唑啉类化合物进行构效关系分析时发现, 喹唑啉3位上的N并不直接与酶的残基结合, 将该N原子换成C并引入吸电子基团时并不影响化合物的活性。在喹啉环C6和C7引入甲氧基或其他含氧基团, C4位引入氧取代基, 是最具代表性的骨架。
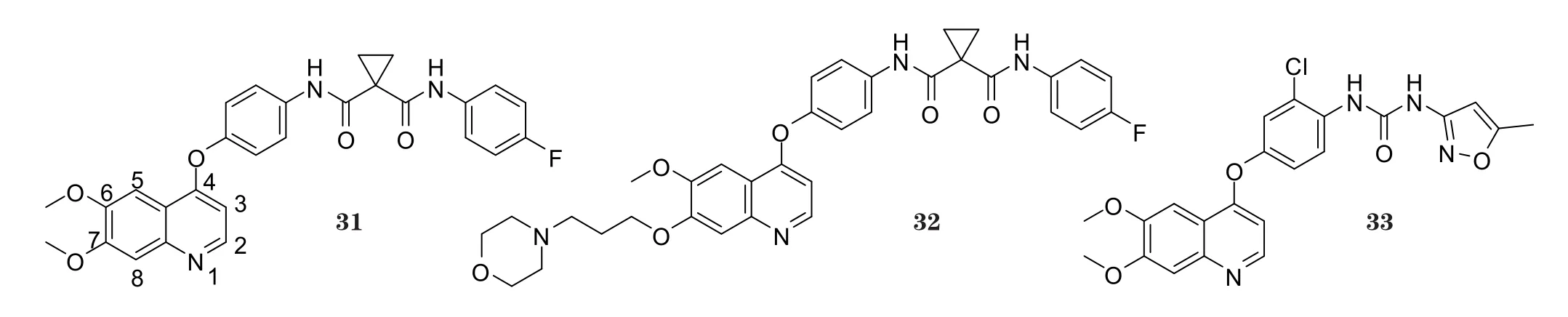
Cabozantinib(XL184, BMS-907351, 31)是由Exelixis公司研制开发的口服小分子多靶点酪氨酸激酶抑制剂,于2012年11月获FDA批准用于治疗不可手术切除的晚期局部恶性肿瘤或转移性甲状腺髓样癌(MTC)[22]。Foretinib(GSK1363089, XL880, 32)是由葛兰素史克公司开发的多靶点酪氨酸激酶抑制剂, 对VEGFR具有特异性抑制作用(IC50为6.8 nmol·L-1), 目前正在进行有关foretinib治疗恶性实体瘤的Ⅰ/Ⅱ期临床研究[23]。Tivozanib(KRN951, AV-951, 33)是由美国AEVO制药公司开发的VEGFR-2选择性抑制剂(其对VEGFR-2的体外IC50为6.5 nmol·L-1), 其目前正在进行Ⅲ期临床研究(Nakamura K, Cancer Res, 2006年)。
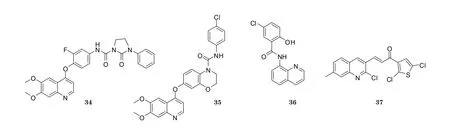
c-Met是一种由原癌基因c-Met编码的蛋白产物, 其为肝细胞生长因子受体, 具有酪氨酸激酶活性, 与肿瘤的生长、转移密切相关。近年来一系列以c-Met和VEGFR-2为靶点的双重抑制剂已成为喹啉类激酶抑制剂开发的新方向。例如, 化合物34对c-Met和VEGFR-2的IC50均在纳摩尔级, 对人类肿瘤细胞株具有抗增殖作用[24]。化合物35对VEGFR-2和HUVEC均具有抑制作用, 体外IC50分别为0.5和4 nmol·L-1(La D S, J Med Chem, 2008年)。
1.4.2 非稠合吡啶类 瓦他拉尼(vatalanib, PTK-787, ZK222584, 38)是诺华公司和先灵葆雅公司联合开发的小分子VEGFR酪氨酸激酶抑制剂, 目前正处于Ⅲ期临床研究阶段。Telatinib(BAY57-9352, 39)是拜耳公司研发的口服小分子酪氨酸激酶抑制剂, 其母核特征是具有呋喃并哒嗪结构(Geiderblom H, Eur J Cancer, 2006年)。Telatinib对VEGFR-2和VEGFR-3均具有抑制活性, 体外IC50分别为6和4 nmol·L-1。体内研究表明, telatinib可通过阻断以酰胺键代替氧连接喹啉环与苯环所得的衍生物中, 化合物36对VEGFR-2和HUVEC有良好的抑制活性(体外IC50分别为3.8和5.5 nmol·L-1)[25]。化合物37的噻吩环中, 2位氯与Lys868形成氢键, 5位氯与Asp1046形成氢键,从而使化合物与VEGFR-2激酶的ATP口袋的结合能力更强。化合物37对VEGFR-2和HUVEC均具有良好的抑制活性, 其体外IC50分别为73.41和21.78 nmol·L-1[26]。VEGFR信号通路和肿瘤血管生成而抑制肿瘤生长和转移。研究显示, telatinib的吡啶甲酰胺的羰基氧能与VEGFR-2的Cys917直接形成氢键。苯胺部分位于疏水区, 而酞嗪母根据vatalanib及其他酞嗪类抑制剂的结构特征和空间构象研究结果, 诺华公司设计并合成了一系列邻氨基苯甲酰衍生物, 其中, 化合物AAL-993(40)上的NH键和羰基形成伪环, 其对VEGFR-2具有抑制活性, IC50为23 nmol·L-1(Manley P W, J Med Chem, 2002年)。化合物41以异噻唑环代替苯环, 化合物42以S原子取代NH, 这2个化合物对VEGFR-2的IC50分别为41和26 nmol·L-1[27]。核与氨基酸残基 Val914、Val865和Cys1043可产生疏水作用(见图2)。含三氮唑环的化合物43, 其体外抑酶活性和抗肿瘤细胞增殖活性与vatalanib相当[28]。化合物44对VEGFR-2具有抑制作用, 其IC50为24 nmol·L-1。研究显示, 该化合物苯环上的F能与Leu788、Met766和Glu762之间形成分子间作用力。化合物44中吡啶环具有增加水溶性的作用;化合物的氨基未被取代时, 在苯环4位引入电负性高的卤素原子,在3位引入电负性低的卤素原子, 化合物活性增强[29]。


图2 Telatinib与VEGFR-2的结合模式图Figure 2 The model of telatinib binding with VEGFR-2

1.5 其他类
阿西替尼(axitinib, 45)是由辉瑞公司开发的吲唑类多激酶抑制剂, 其对VEGFR-1、VEGFR-2、VEGFR-3、PDGFR和c-Kit等均有抑制作用, 该化合物已于2012年1月获FDA批准用于治疗晚期肾癌[30]。吡咯三嗪衍生物46具有显著的VEGFR-2抑制活性, 其对VEGFR-2和HUVEC的IC50分别为11和25 nmol·L-1(Cai Z W, Bioorg Med Chem Lett, 2008年)。二唑环上连有对氧基六氢吡啶等脂肪杂环时, 可增加化合物的水溶性从而提高其抑制活性。BMS-645737(47)对VEGFR-2的体外IC50为25 nmol·L-1, 该化合物对人体肿瘤异种移植模型亦具有良好的体内抑制活性(Ruel R, Bioorg Med Chem Lett, 2008年)。
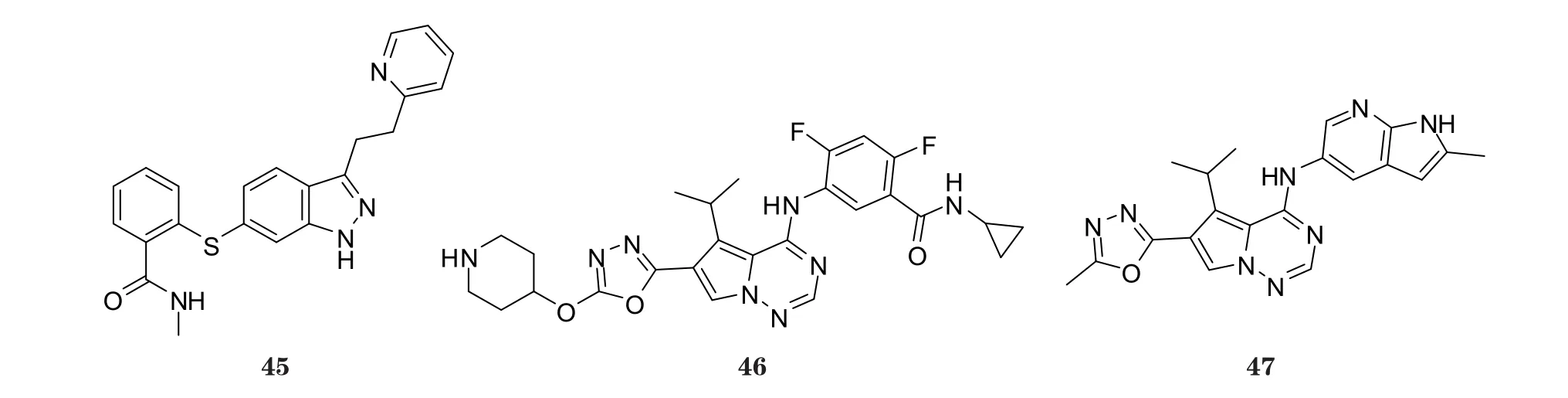
四取代的吡唑类衍生物48是一种多效激酶抑制剂,该化合物吡啶环上的N与Cys919形成一个氢键, 其三氯苯基作为一个疏水区与Thr916和Lys868产生分子间作用;当苯环4位取代基为三氯苯基等吸电子基团时, 化合物活性增强。本品对Src、野生型B-Raf、B-RafV600E、EGFR和VEGFR-2均具有一定的抑制作用[31]。化合物49对VEGFR-2和HUVEC均具有抑制作用(体外IC50分别为2和5 nmol·L-1), 该化合物母核为三唑并吡啶, 三唑环上的1位N原子与Cys919形成关键的氢键, 化合物的吡唑部分构成一个疏水区, 吡唑环连接的羰基与Asp1046形成一个氢键。构效关系研究显示, 苯环上有卤素取代时, 化合物活性比烷基取代时更强[32]。
化合物50是一种选择性的VEGFR-2抑制剂(其对VEGFR-2体外IC50为13 nmol·L-1), 该化合物嘧啶环上的1位N原子和吲哚环上9位NH能与Cys917形成氢键, 化合物2位氨基可以和Glu915形成氢键, 而吲哚环与 Val914、Phe916、Cys917、Leu1033、Cys1043、Leu838和Val846有疏水作用, 苯胺上的苯环和Val846、Ala864、Lys866、Val897、Val914及Leu1033产生疏水作用。研究发现, 苯胺环上3位有取代的化合物对VEGFR-2的抑制活性低于4位和2位有取代时的活性, 苯环上有卤素取代时比有烷基取代时的活性高[33]。
化合物BMS-582664(51)是由百时美施贵宝公司研发的口服小分子多靶点酪氨酸激酶抑制剂, 是ATP竞争性的VEGFR-2抑制剂, 其体外IC50为25 nmol·L-1。该化合物对分别荷有肺癌L2987和结肠癌HCT116细胞的小鼠均显示出抑制肿瘤血管内皮细胞增长的活性。目前正在进行将BMS-582664用于治疗肝癌的Ⅲ期临床研究, 然而结果显示, 与接受标准疗法的肝癌患者相比, 该化合物未能提高患者的总生存率。多靶点酪氨酸激酶抑制剂——化合物52是吡咯并嘧啶的衍生物, 其对VEGFR-2的IC50为27 nmol·L-1, 该化合物对人胃癌GTL-16细胞具有增殖抑制作用[34]。
吡啶并三嗪类衍生物53在质量浓度为10 mg·L-1时对VEGFR-2抑制率为40%。化合物中苯胺基团与Lys866、Val897、Leu887、Val912、Val914、Cys1043以及Phe1045间有疏水作用;化合物的NH与Cys917之间有氢键作用;吡啶并三嗪母核与Leu838、Gly920及Leu1033间有疏水作用, 增强了化合物与VEGFR-2的结合能力[35]。

2 不可逆抑制剂
近年来, 不可逆型酪氨酸激酶抑制剂的研究受到越来越多的关注[36-37]。不可逆型抑制剂是含有活性官能团且能与靶蛋白特异性氨基酸残基反应并形成共价键的化合物。不可逆型酪氨酸激酶抑制剂的设计思路在于, 受体蛋白中均含有半胱氨酸残基, 而其他激酶中的相应位置则是谷氨酸或丝氨酸残基, 因此, 在小分子抑制剂的结构中引入能和半胱氨酸的巯基形成共价键的基团可以实现对酪氨酸激酶的不可逆抑制。不可逆激酶抑制剂具有高选择性、高活性、作用时间长、给药剂量小以及可对抗耐药肿瘤等潜在优势。
现有的不可逆抑制剂中, 针对EGFR的不可逆抑制剂的研究较多[38], 而VEGFR的不可逆抑制剂研究较少。文献报道的不可逆VEGFR抑制剂中均含有活性醌结构, 化合物54中的苯醌环可通过氧化还原反应与ATP结合位点的Cys1045发生共价结合, 该化合物对VEGFR-2具有抑制作用, 体外IC50达纳摩尔水平;在荷有结肠癌DLD1的裸鼠中亦表现出显著的抗肿瘤活性[39]。Hypothemycin(55)是由圣克鲁斯公司研发的共价酶抑制剂, 其结构中包含顺-烯酮迈克尔受体, 从而能与Cys1045共价结合, 该化合物对MEK1/2、ERK1/2、PDGFR、FLT3和VEGFR均有抑制作用。Hypothemycin的酚羟基与酶铰链段的Met108形成2个氢键, 其烯酮结构对硫醇基团敏感, 提高了其对VEGFR-2的选择性。动物实验表明, 由hypothemycin衍生的化合物56和57表现出良好的耐药性和稳定性;化合物56抑制血管生成的能力以及化合物57抑制肿瘤细胞迁移的活性均优于舒尼替尼[40]。

化合物58和59中亲电性的苯醌基团可与Cys1045共价结合, 喹唑啉N1与Cys919能形成关键的氢键。苯醌的4位取代基为苄氧基时, 取代基上的苯环与Phe1047可形成分子间作用, 使化合物对VEGFR-2的抑制作用加强;当取代基为其他烷氧基时, 化合物抑制活性下降。化合物59中烯酮迈克尔受体与Cys733发生共价结合。化合物58和59对 VEGFR-2均具有抑制作用, 其IC50分别为50和102 nmol·L-1[41-42]。

3 结语
以抑制VEGFR为基础的抗血管生成疗法已成为癌症治疗中最有效的手段之一。近几年, 研究设计和合成小分子VEGFR抑制剂也取得了一定成果, 许多高效低毒的具有前景的化合物已进入临床研究阶段。随着对不可逆型抑制剂的开发以及对VEGF/VEGFR信号通路和作用机制的深入理解, 越来越多的小分子VEGFR抑制剂将会不断涌现, 成为人类对抗肿瘤强有力的武器。在不久的将来, VEGFR抑制剂的研究重点可能会转向不可逆型抑制剂, 部分药物选择性不高、易引发耐药性等问题亦有望获得解决。笔者所在课题组正在开展基于二芳基脲结构的不可逆酪氨酸激酶抑制剂的研究, 以现有的非共价抑制剂结构为基础,希望通过合理引入可与靶标结构中特定蛋白结合的共价结合基团, 设计合成新型多靶点的不可逆酪氨酸激酶抑制剂,目前合成工作正在进行中。
[1]Smith N R, Baker D, James N H, et al. Vascular endothelial growth factor receptors VEGFR-2 and VEGFR-3 are localized primarily to the vasculature in human primary solid cancers [J]. Clin Cancer Res, 2010, 16 (14): 3548-3561.
[2]Yakes F M, Chen J, Tan J, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth [J]. Mol Cancer Ther, 2011, 10 (12): 2298-2308.
[3]Qian F, Engst S, Yamaguchi K, et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases [J]. Cancer Res, 2009, 69 (20): 8009-8016.
[4]Roth G J, Heckel A, Colbatzky F, et al. Design, synthesis, and evaluation of indolinones as triple angiokinase inhibitors and the discovery of a highly specific 6-methoxycarbonyl-substituted indolinone (BIBF 1120) [J]. J Med Chem, 2009, 52 (14): 4466-4480.
[5]Chiu Y L, Carlson D M, Pradhan R S, et al. Exposure-response (safety) analysis to identify linifanib dose for a phase III study in patients with hepatocellular carcinoma [J]. Clin Ther, 2013, 35 (11): 1770-1777.
[6]Oguro Y, Miyamoto N, Okada K, et al. Design, synthesis, and evaluation of 5-methyl-4-phenoxy-5H-pyrrolo [3, 2-d] pyrimidine derivatives: novel VEGFR2 kinase inhibitors binding to inactive kinase conformation [J]. Bioorg Med Chem, 2010, 18 (20): 7260-7273.
[7]Oguro Y, Miyamoto N, Okada K, et al. N-Phenyl-N′-[4-(5H-pyrrolo [3, 2-d] pyrimidin-4-yloxy) phenyl] ureas as novel inhibitors of VEGFR and FGFR kinases [J]. Bioorg Med Chem, 2010, 18 (20): 7150-7163.
[8]Sanphanya K, Wattanapitayakul S K, Phowichit S, et al. Novel VEGFR-2 kinase inhibitors identified by the back-to-front approach[J]. Bioorg Med Chem Lett, 2013, 23 (10): 2962-2967.
[9]Lin W H, Hsu J T, Hsieh S Y, et al. Discovery of 3-phenyl-1H-5-pyrazolylamine derivatives containing a urea pharmacophore as potent and efficacious inhibitors of FMS-like tyrosine kinase-3 (FLT3) [J]. Bioorg Med Chem, 2013, 21 (11): 2856-2867.
[10]Girard N. Antiangiogenic agents and chemotherapy in advanced nonsmall cell lung cancer: a clinical perspective [J]. Expert Rev Anticancer Ther, 2013, 13 (10): 1193-1206.
[11]Wang D, Tang F, Wang S, et al. Preclinical anti-angiogenesis and anti-tumor activity of SIM010603, an oral, multitargets receptor tyrosine kinases inhibitor[ J]. Cancer Chemother Pharmacol, 2012, 69 (1): 173-183.
[12]Cho T P, Dong S Y, Jun F, et al. Novel potent orally active multitargeted receptor tyrosine kinase inhibitors: synthesis, structureactivity relationships, and antitumor activities of 2-indolinone derivatives[J]. J Med Chem, 2010, 53 (22): 8140-8149.
[13]Hudkins R L, Zulli A L, Underiner T L, et al. 8-THP-DHI analogs as potent type I dual TIE-2/VEGF-R2 receptor tyrosine kinase inhibitors[J]. Bioorg Med Chem Lett, 2010, 20 (11): 3356-3360.
[14]Kim Y S, Li F, O’Neill B E, et al. Specific binding of modified ZD6474 (vandetanib) monomer and its dimer with VEGF receptor-2[ J]. Bioconjug Chem, 2013, 24 (11): 1937-1944.
[15]Takeda H, Takigawa N, Ohashi K, et al. Vandetanib is effective in EGFR-mutant lung cancer cells with PTEN deficiency [J]. Exp Cell Res, 2013, 319 (4): 417-423.
[16]Yu B, Tang L D, Li Y L, et al. Design, synthesis and antitumor activity of 4-aminoquinazoline derivatives targeting VEGFR-2 tyrosine kinase[J]. Bioorg Med Chem Lett, 2012, 22 (1): 110-114.
[17]Zhang S, Cao Z, Tian H, et al. SKLB1002, a novel potent inhibitor of VEGF receptor 2 signaling, inhibits angiogenesis and tumor growth in vivo [J]. Clin Cancer Res, 2011, 17 (13): 4439-4450.
[18]Plé P A, Jung F, Ashton S, et al. Discovery of AZD2932, a new quinazoline ether inhibitor with high affinity for VEGFR-2 and PDGFR tyrosine kinases [J] Bioorg Med Chem Lett, 2012, 22 (1): 262-266.
[19]Conconi M T, Marzaro G, Urbani L, et al. Quinazoline-based multi-tyrosine kinase inhibitors: synthesis, modeling, anti tumor and antiangiogenic properties [J]. Eur J Med Chem, 2013, 67 (9): 373-383.
[20]Yakes F M, Chen J, Tan J, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth [J]. Mol Cancer Ther, 2011, 10 (12):2298-2308.
[21]Choi H E, Choi J H, Lee J Y, et al. Synthesis and evaluation of nicotinamide derivative as antiangiogenic agents [J]. Bioorg Med Chem Lett, 2013, 23 (7): 2083-2088.
[22]Yakes F M, Chen J, Tan J, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth [J]. Mol Cancer Ther, 2011, 10 (12): 2298-2308.
[23]Qian F, Engst S, Yamaguchi K, et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases [J].
[24]Cancer Res, 2009, 69 (20): 8009-8016. Mannion M, Raeppel S, Claridge S, et al. N-(4-(6, 7-disubstitutedquinolin-4-yloxy)-3-fluorophenyl)-2-oxo-3-phenylimidazolidine-1-carboxamides: a novel series of dual c-Met/VEGFR2 receptor tyrosine kinase inhibitors [J]. Bioorg Med Chem Lett, 2009, 19 (23):
[25]6552-6556. Yang Y, Shi L, Zhou Y, et al. Design, synthesis and biological evaluation of quinoline amide derivatives as novel VEGFR-2 inhibitors
[26][J]. Bioorg Med Chem Lett, 2010, 20 (22): 6653-6656. Rizvi S U, Siddiqui H L, Nisar M, et al. Discovery and molecular docking of quinolyl-thienyl chalcones as antiangiogenic agents targeting VEGFR-2 tyrosine kinase [J]. Bioorg Med Chem Lett, 2012, 22 (2):
[27]942-944. Kiselyov A S, Semenova M, Semenov V V. 3, 4-Disubstituted isothiazoles: novel potent inhibitors of VEGF receptors 1 and 2 [J].
[28]Bioorg Med Chem Lett, 2009, 19 (4): 1195-1198. Kiselyov A S, Semenova M, Semenov V V. (1, 2, 3-Triazol-4-yl) benzenamines: synthesis and activity against VEGF receptors 1 and 2[ J].
[29]Bioorg Med Chem Lett, 2009, 19 (5): 1344-1348. Gangjee A, Namjoshi O A, Yu J, et al. N2-Trimethylacetyl substituted and unsubstituted-N4-phenylsubstituted-6-(2-pyridin-2-ylethyl)-7H-pyrrolo [2, 3-d] pyrimidine-2, 4-diamines: design, cellular receptor tyrosine kinase inhibitory activities and in vivo evaluation as antiangiogenic, antimetastatic and antitumoragents [J]. Bioorg Med Chem, 2013, 21 (5): 1312-1323.
[30]Ho T H, Jonasch E. Axitinib in the treatment of metastatic renal cell carcinoma [J]. Future Oncol, 2011, 7 (11): 1247-1253.
[31]Abu Thaher B, Arnsmann M, Totzke F, et al. Tri- and tetrasubstituted pyrazole derivates: regioisomerism switches activity from p38MAP kinase to important cancer kinases [J]. J Med Chem, 2012, 55 (2): 961-965.
[32]Oguro Y, Cary D R, Miyamoto N, et al. Design, synthesis, and evaluation of novel VEGFR2 kinase inhibitors: discovery of [1, 2, 4] triazolo [1, 5-a] pyridine derivatives with slow dissociation with slow dissociation kinetics [J]. Bioorg Med Chem, 2013, 21 (15): 4714-4729.
[33]Gangjee A, Zaware N, Raghavan S, et al. Synthesis and biological activity of 5-chloro-N4-substituted phenyl-9H-pyrimido [4, 5-b] indole-2, 4-diamines as vascular endothelial growth factor receptor-2 inhibitors and antiangiogenic agents[ J]. Bioorg Med Chem, 2013, 21 (7): 1857-1864.
[34]Zhao F, Derbin G, Miller S, et al. Acid-catalyzed hydrolysis of BMS-582664: degradation product identification and mechanism elucidation[J]. J Pharm Sci, 2012, 101 (9): 3526-3530.
[35]Zhao X W, Liu D, Luan S L, et al. Synthesis and biological evaluation of substituted 1, 2, 3-benzotriazines and pyrido [3, 2-d]-1, 2, 3-triazines as inhibitors of vascular endothelial growth factor receptor-2[J]. Bioorg Med Chem, 2013, 21 (24): 7807-7815.
[36]Liu Q, Sabnis Y, Zhao Z, et al. Developing irreversible inhibitors of the protein kinase cysteinome [J]. Chem Biol, 2013, 20 (2): 146-159.
[37]Carmi C, Mor M, Petronini P G, et al. Clinical perspectives for irreversible tyrosine kinase inhibitors in cancer [J]. Biochem Pharmacol, 2012, 84 (11): 1388-1399.
[38]Zhou W, Ercan D, Chen L, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M[J]. Nature, 2009, 462 (7276): 1070-1074.
[39]Wissner A, Floyd M B, Johnson B D, et al. 2-(Quinazolin-4-ylamino)-[1, 4] benzoquinones as covalent-binding, irreversible inhibitors of the kinase domain of vascular endothelial growth factor receptor-2 [J]. J Med Chem, 2005, 48 (24): 7560-7581.
[40]Barluenga S, Jogireddy R, Koripelly G K, et al. In vivo efficacy of natural product-inspired irreversible kinase inhibitors [J]. Chembiochem, 2010, 11 (12): 1692-1699.
[41]Wissner A, Floyd M B, Johnson B D, et al. 2-(Quinazolin-4-ylamino)-[1, 4] benzoquinones as covalent-binding, irreversible inhibitors of the kinase domain of vascular endothelial growth factor receptor-2 [J]. J Med Chem, 2005, 48 (24): 7560-7581.
[42]Wissner A, Fraser H L, Ingalls C L, et al. Dual irreversible kinase inhibitors: quinazoline-based inhibitors incorporating two independent reactive centers with each targeting different cysteine residues in the kinase domains of EGFR and VEGFR-2 [J]. Bioorg Med Chem, 2007, 15 (11): 3635-3648.
Advances in Research on VEGFR-2 Tyrosine Kinase Inhibitors
YE Yanzhao, SHI Zhihao, LU Tao
(School of Science, China Pharmaceutical University, Nanjing 211198, China)
Vascular endothelial growth factor receptor (VEGFR) is a kind of important tyrosine kinase, which can induce angiogenesis. It can be found overexpressed in tumor tissues. Recently, the VEGFR-based anti-angiogenic therapy has become a new cancer treatment strategy. More and more small-molecule inhibitors of VEGFR-2 have been discovered. The design and synthesis of VEGFR-2 inhibitors have also drawn a wide public attention. The advances in research on VEGFR-2 inhibitors in the past 5 years have been reviewed in this paper.
antitumor; angiogenesis inhibitor; VEGFR-2; tyrosine kinase inhibitor其中VEGFR-2是一种特异性糖蛋白, 相对分子质量为210 000~230 000, 主要分布在血管内皮细胞和造血干细胞中, 可以与VEGF-A、VEGF-C、VEGF-D、VEGF-E结合,主要调节VEGF在血管内皮细胞中的生理反应, 包括通透性、增殖和迁移, 是生理性和病理性血管生成过程中的一个关键信号传感器。VEGFR-2在卵巢癌、甲状腺癌、黑色素瘤和髓母细胞瘤中过度表达, 主要通过调控肿瘤脉管系统(包括血液和淋巴)给大部分肿瘤组织提供营养。此外, VEGFR-2在恶性肠癌、肺癌、乳腺癌等肿瘤中的表达水平亦显著高于非肿瘤组织[1]。
R979.1
A
1001-5094 (2014) 01-0014-11
接受日期:2013-11-05
项目资助:国家自然科学基金资助项目(No.21302225);*
陆涛,教授;
研究方向:药物分子的设计与合成;
Tel:025-86185086;E-mail:lutao@cpu.edu.cn
猜你喜欢
高中数理化(2022年14期)2022-08-15
波谱学杂志(2021年3期)2021-09-07
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
原子与分子物理学报(2019年5期)2019-04-28
中成药(2018年12期)2018-12-29
天然产物研究与开发(2018年1期)2018-02-02
中成药(2018年1期)2018-02-02
中国塑料(2016年7期)2016-04-16
中学化学(2015年12期)2016-01-19
