
多重靶向的4-取代苯胺喹唑啉类衍生物抗肿瘤活性研究进展
2014-03-14 09:48龚飞虎孙丽萍
药学进展 2014年1期
龚飞虎, 孙丽萍
(中国药科大学药物化学教研室, 江苏 南京 210009)
多重靶向的4-取代苯胺喹唑啉类衍生物抗肿瘤活性研究进展
龚飞虎, 孙丽萍*
(中国药科大学药物化学教研室, 江苏 南京 210009)
4-取代苯胺喹唑啉类化合物是喹唑啉类酪氨酸激酶抑制剂中活性较高的一类化合物, 其对表皮生长因子受体、血管内皮生长因子受体、组蛋白去乙酰化酶等均有抑制作用, 已成为抗肿瘤药物的研究热点。综述了近几年多重靶向的4-取代苯胺喹唑啉类化合物抗肿瘤活性的研究进展。
4-取代苯胺喹唑啉;人类表皮生长因子受体;血管内皮生长因子受体;组蛋白去乙酰化酶;多靶向;抗肿瘤活性
恶性肿瘤是严重威胁人类生命的疾病, 其发生机制是基因表达异常引起细胞信号传导通路的失调, 导致细胞无限增殖, 其中酪氨酸激酶(TK)起到关键作用[1]。自1994年Fry等[2]发现4-苯胺基喹唑啉(PD153035)可用作EGFR酪氨酸激酶的特异性抑制剂以来, 喹唑啉类化合物成为酪氨酸激酶抑制剂类抗肿瘤药研发的热点之一, 目前也已有不少关于喹唑啉类抗肿瘤活性化合物的综述报道[3-5]。其中, 4-取代苯胺喹唑啉类化合物(其喹唑啉母核见结构式1)是喹唑啉类酪氨酸激酶抑制剂中活性较高的一类化合物, 能同时靶向表皮生长因子受体(EGFR)/ ErbB-2、EGFR/血管内皮生长因子受体-2(VEGFR-2)、EGFR/VEGFR-2/组蛋白去乙酰化酶(HDAC)等多种靶点的4-苯胺喹唑啉类化合物成为研究的热点。本文介绍了近几年多重靶点的4-取代苯胺喹唑啉类化合物抗肿瘤活性的研究进展。

1 对EGFR/ErbB-2有双重抑制作用的4-取代苯胺喹唑啉类化合物
表皮生长因子受体酪氨酸激酶类家族包括EGFR/ ErbB-1、HER2/ErbB-2、HER3/ErbB-3、HER4/ErbB-4, 广泛分布于哺乳动物的上皮细胞膜上, 其信号传导途径在癌细胞的增殖、存活和迁移中起到重要作用[6]。靶向EGFR的4-取代苯胺喹唑啉类药物是肿瘤治疗研究的热点之一。化合
F物2~7为近年来设计合成的选择性单靶向EGFR的4-取代苯胺喹唑啉类化合物。Ban等[7]合成了化合物2~4, 其对EGFR的体外IC50分别为2.53、3.16和4.45 nmol·L-1;而阳性对照药吉非替尼(gefitinib)对EGFR的 IC50为21.0 nmol·L-1。2011年, Li等[8]报道了化合物5和6, 其对EGFR过度表达的乳腺癌SK-BR-3细胞和MDA-MB-468细胞的体外IC50分别为9.74和14.19 μmol·L-1, 而对EGFR非过度表达的结肠癌HCT-116细胞的抑制活性较弱(IC50均大于100 μmol·L-1)。2013年, Xu等[9]设计并合成了化合物7, 其对EGFR的IC50为1.5 μmol·L-1。

单靶向表皮生长因子的4-苯胺基喹唑啉类化合物的构效关系研究显示:1)在喹唑啉母核的6、7位引入供电子取代基时能增强喹唑啉母核N1、N3与EGFR激酶的相互作用;2)4位苯胺可与EGFR激酶的疏水口袋相互作用,苯环上不同位置取代能影响化合物对EGFR激酶的选择性。虽然单一靶向EGFR的酪氨酸激酶抑制剂(第1代)具有较好的抗肿瘤活性, 但很容易引发耐药性, 如吉非替尼[10]。研究显示, 在4-取代苯胺喹唑啉类化合物的6、7位引入适当吸电子取代基, 或在4位苯胺上引入脂溶性取代基,能提高该类化合物对EGFR家族的多重抑制作用, 从而避免了耐药性的产生[11]。由此, 第2代EGFR/ErbB-2酪氨酸激酶抑制剂随之而生, 例如lapatinib(8)已于2007年12月获欧洲药品管理局(EMEA)批准上市, 用于治疗晚期或转移性乳腺癌[12]。
Afitinib(9)由勃林格殷格翰公司开发, 其在喹唑啉母核6位引入了具有吸电子作用的酰胺取代基团, 是高选择性的4-取代苯胺喹唑啉类EGFR/HER2酪氨酸激酶的不可逆抑制剂。Yap等[13]通过一项Ⅱ期临床研究评价了afitinib治疗非小细胞肺癌的有效性和安全性, 结果表明, 本品推荐剂量为50 mg·d-1, 研究中最主要的不良反应为胃肠道不适(包括腹泻、恶心、呕吐)。2013年7月, FDA批准afitinib用于EGFR突变阳性的局部晚期或转移性NSCLC的治疗。Zhang等[14]在6位引入具有强吸电子能力的二硫代氨基甲酸酯, 合成了一系列新的4-苯胺喹唑啉类衍生物。其中化合物10具有双重EGFR/ErbB-2抑制作用, 且活性最强, 其对EGFR过度表达的MDA-MB-468细胞和ErbB-2过度表达的SK-BR-3细胞具有显著抑制活性, 体外IC50分别为2.71和 2.62 μmol·L-1, 强于阳性对照药lapatinib(其对上述2种细胞的体外IC50分别为23.46和4.34 μmol·L-1)。


Li等[15]在喹唑啉母核6位引入了具有吸电子作用的水杨酰基, 进而合成了18种该系列的4-取代苯胺喹唑啉类衍生物。其中化合物11具有最强的EGFR/ErbB-2抑制活性, 其IC50分别为0.12和0.096 μmol·L-1, 与阳性对照药埃罗替尼(erlotinib)和lapatinib活性相当。课题组设计的其他所有化合物对A431、MCF-7和A549这3种细胞株的体外IC50为0.49~2.37 μmol·L-1, 活性亦与阳性对照药埃罗替尼(IC50为0.40~1.07 μmol·L-1)和lapatinib(IC50为0.21~1.12 μmol·L-1) 相当。

Barlaam B等[16]发现一类新的高效的EGFR/ErbB-2不可逆抑制剂, 其中AZD8931(12)活性最强, 其在体外抑制EGFR和ErbB-2酪氨酸激酶的IC50分别为14和12 nmol·L-1。在KB细胞中进行的研究显示, AZD8931抑制EGF诱导的EGFR磷酸化的IC50为4 nmol·L-1。在MCF-7细胞中, AZD8931抑制神经生长因子刺激的ErbB-2磷酸化的IC50为3 nmol·L-1。药物分子结构中具有吸电子作用的甲基乙酰化侧链能增强药物对ErbB-2的抑制活性。此外, AZD8931还具有靶向ErbB-3的抑制活性。目前, AZD8931已进入Ⅰ期临床研究, 用于治疗晚期实体瘤患者[17]。除了在喹唑啉母核的6位上引入吸电子取代基以外,研究人员还尝试了其他类型的修饰方式。Ahmed等[18]在4位苯胺上引入了脂溶性基团, 合成了一类新的4-取代苯胺喹唑啉类EGFR/ErbB-2酪氨酸激酶抑制剂衍生物。其中,活性最强的化合物13对EGFR/ErbB-2的体外IC50分别为1.935和1.035 μmol·L-1。这些新的4-取代苯胺喹唑啉类衍生物在EGFR/ErbB-2酶键合位点处能够与额外的氨基酸残基发生结合(即能与EGFR上的Asp855和Lys745,以及与ErbB-2上的Asp863和Lys753通过氢键发生相互作用), 降低键合能, 有利于化合物与激酶之间的相互作用。


理论上认为, 第2代EGFR不可逆抑制剂相比于第1代EGFR可逆抑制剂的优点主要包括3个方面:1)第2代抑制剂对ErbB酪氨酸激酶信号传导的抑制作用更持久;2)第2代抑制剂具有更完整的EGFR信号通路阻断作用;3)在针对某些突变型肿瘤方面, 第2代抑制剂明显优于第1代抑制剂, 例如第2代抑制剂可有效地抑制T790M突变型或其他稀有突变型的非小细胞肺癌的增殖, 而第1代EGFR抑制剂则无效[19]。开发可双重靶向EGFR/ErbB-2的新化合物已成为酪氨酸激酶抑制剂研究热点。
2 对EGFR/VEGFR-2具有多重抑制作用的4-取代苯胺喹唑啉类化合物
肿瘤血管是异常增生的血管。VEGF及其受体VEGFR是肿瘤血管生成最重要的靶分子, 也是迄今为止研究最多且最深入的肿瘤新生血管标志分子。VEGFR-2被视为抑制肿瘤血管形成的最重要的靶向分子靶点[20]。单一靶点的小分子药物的治疗范围窄, 难以有效阻断与肿瘤细胞发生、发展相关的信号通路, 而具有多靶向机制的药物则有望克服这一困难, 现已成为新的研究趋势[21]。
目前已上市的药物伐地他尼(vandetanib, 14)是阿斯利康公司开发的一种4-取代苯胺喹唑啉类多靶点酪氨酸激酶抑制剂, 其对VEGFR-1、VEGFR-2、EGFR、FGFR-1的体外IC50分别为1.6、0.04、0.5和3.6 μmol·L-1。2005年10月, 本品获得了罕见病用药资格, 所针对的适应证为滤泡型、髓质型、未分化型以及局部复发或转移的乳突型甲状腺癌[5]。2011年8月, 伐地他尼获FDA批准, 成为首个用于治疗晚期甲状腺髓样癌的药物。
除了伐地他尼外, 近几年研究人员针对EGFR/VEGFR还设计合成了其他一些4-取代苯胺喹唑啉类化合物。例如, Nakamura等[22]设计合成了一系列在喹唑啉母核或苯胺对位有硼酸取代的4-取代苯胺喹唑啉类EGFR/VEGFR酪氨酸激酶抑制剂。构效关系研究显示, 当硼酸酯或硼酸取代基在6位时, 化合物对EGFR的抑制活性较好, 如化合物15对EGFR的IC50为0.46 μmol·L-1;当硼酸酯或硼酸取代基在4位氨基苯环上, 化合物对VEGFR-2具有显著抑制活性, 如化合物16对VEGFR-2的IC50为0.036 μmol·L-1, 活性与阳性对照药AAL993(其对VEGFR-2的IC50为0.014 μmol·L-1)相当。

Garofalo等[23]合成了3类在4位上分别有不同取代基团(苯胺、N-甲基苯胺、芳氧基)的4-取代苯胺喹唑啉类衍生物。构效关系研究显示, 4位为苯胺取代的化合物17对EGFR和VEGFR-2的IC50均高于5 μmol·L-1, 而4位为芳氧基的化合物18对VEGFR-2抑制活性较高(IC50为0.7 μmol·L-1), 对EGFR抑制活性较弱(IC50高于10 μmol·L-1)。

Conconi等[24]合成了在喹唑啉母核上并入苯环或含氧杂环的一类新的4-苯胺喹唑啉类多靶点酪氨酸激酶抑制剂。其中化合物19具有显著的抗肿瘤细胞增殖活性, 其对A431细胞的IC50为0.81 μmol·L-1, 对MCF-7β的IC50为0.77 μmol·L-1, 此外对EGFR、FGFR-1、VEGFR-2、 PDGFRβ、Src、Abl等酪氨酸激酶亦具有较强的抑制活性, IC50分别为0.002、0.063、0.848、0.243、0.032和0.051 μmol·L-1。化合物19对人脐静脉血管内皮细胞(HUVEC)的体外IC50为0.75 μmol·L-1。 Zhang等[25]分别在4-苯胺喹唑啉母核的不同位置引入苯脲基团, 成功地设计合成了一类新型多靶点激酶抑制剂。其中, 化合物20对VEGFR-2的体外IC50为17 nmol·L-1(阳性对照sorafenib对VEGFR-2的体外IC50为52 nmol·L-1), 对EGFR的IC50为285 nmol·L-1。化合物21具有显著的EGFR抑制活性(IC50为82 nmol·L-1), 而对VEGFR-2的抑制活性一般, IC50为213 nmol·L-1。由此可见, 二苯脲基在4-苯胺的苯环上的取代位置对该类化合物的EGFR/VEGFR-2选择性具有重要影响。


3 对EGFR /VEGFR-2/ HDAC具有多重抑制作用的4-取代苯胺喹唑啉类化合物
细胞增殖与细胞周期的运转有密切的关系, 调控细胞周期可以影响到细胞增殖。HDAC抑制剂对细胞周期进程的阻断作用对于该类化合物发挥抑制细胞增殖的效应具有非常重要的意义[26]。多重靶向EGFR、VEGFR-2、HDAC的4-苯胺喹唑啉类化合物可以从抑制肿瘤细胞信号传导途径、抑制肿瘤血管生成、阻断肿瘤细胞周期进程等多方面抑制肿瘤的形成、生长和迁移。近几年研究人员利用拼接方法将HDAC抑制剂的活性药效团引入到4-苯胺喹唑啉类化合物的结构中, 从而设计出多重靶向EGFR、VEGFR-2、HDAC的新颖化合物。
目前研究较多的是在4-取代苯胺喹唑啉类化合物的母核6位引入HDAC抑制剂的活性药效团。Cai等[27]通过将HDAC抑制剂的药效团异羟肟酸拼接到具有EGFR和VEGFR-2抑制剂药效团的4-苯胺喹唑啉类母核上,设计合成了一类具有潜在多重抑制EGFR、VEGFR-2、HDAC活性的化合物。其中, CUDC-101(22)在体外对HDAC、EGFR、VEGFR-2的IC50分别为4.4、2.4和15.7 nmol·L-1。其抗肿瘤细胞增殖活性比阳性对照药voribostat(HDAC抑制剂)、埃罗替尼(EGFR抑制剂)、lapatinib(EGFR/ErbB-2抑制剂)以及联合使用voribostat/埃罗替尼、voribostat/lapatinib的活性更强。CUDC-101目前已进入Ⅰ期临床研究, 用于治疗头颈癌、胃癌、乳腺癌、肝癌和非小细胞肺癌等[28]。

Mahboobi等[29]在喹唑啉母核6位引入HDAC抑制剂药效团, 设计出多靶点抑制HDAC、VEGFR-2、EGFR的小分子化合物。其中, 化合物23对EGFR、VEGFR-2、HDAC的体外IC50分别为0.018、0.011和0.047 μmol·L-1。细胞毒性测定结果显示, 该化合物对多种肿瘤细胞均具有较好的抗增殖活性, 如对HDAC高表达的A549的IC50为1.1~27 μmol·L-1, 对EGFR高表达的Ca127的IC50为0.007~4.0 μmol·L-1, 对VEGFR-2高表达的SKBR3的IC50为0.002~4.4 μmol·L-1。

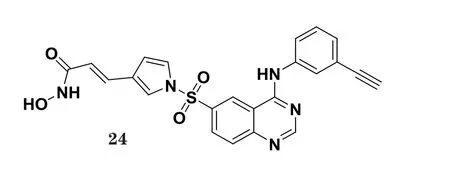
Beckers等[30]将EGFR/VEGFR-2抑制剂埃罗替尼的活性药效团与HDAC抑制剂活性药效团进行拼接, 设计出一类新的HDAC/EGFR/VEGFR-2多靶点抑制剂。其中, 化合物24对EGFR、VEGFR、HDAC的体外IC50分别为1.2、3.4和0.006 4 μmol·L-1(阳性对照SAHA对HDAC的IC50为0.079 μmol·L-1)。此外, 研究人员将HDAC活性药效团引入到4-取代苯胺喹唑啉类化合物的4位苯胺苯环上的适当位置, 也得到了新型多重抑制EGFR、VEGFR-2、HDAC的化合物。Zhang等[31]设计了一类在喹唑啉母核4位苯胺的苯环上接有2个药效团的可靶向EGFR、VEGFR-2、HDAC的4-取代苯胺喹唑啉类化合物, 其中化合物25与阳性对照药伏立诺他(vorinostat)具有相近的HDAC抑制活性, 对HDAC1、HDAC3、HDAC6的IC50分别为0.16、0.18和0.56 μmol·L-1, 对EGFR和VEGFR-2的IC50分别为9.7和47.2 μmol·L-1。

4 展望
本文对近几年来具有双重或多重抑制EGFR、ErbB-2、VEGFR-2、HDAC等活性的4-苯胺喹唑啉类化合物的研究进展进行了简介。可以看出, 对具有抗肿瘤活性的4-苯胺喹唑啉结构的改造, 主要集中于喹唑啉母核的6、7位,或是4位苯胺的苯环上;此外, 活性基团拼接法也是喹唑啉类化合物结构改造的重要方法, 有助于多靶点抗肿瘤活性候选药物的发现。4-苯胺喹唑啉类衍生物表现出显著的EGFR、ErbB-2、VEGFR-2、HDAC抑制活性以及对肿瘤细胞的增殖抑制作用, 且该类化合物有着较大的结构改造空间, 故其作为抗肿瘤药物具有广阔的开发前景。总之,相信在不久的将来会有更多的4-苯胺喹唑啉类抗肿瘤药物出现, 为肿瘤患者带来福音。
[参 考 文 献]
[1]Zwick E, Bange J, Ullrich A. Receptor tyrosine kinase signalling as a target for cancer intervention strategies [J]. Endocr Relat Cancer, 2001, 8 (3): 161-173.
[2]Fry D W, Kraker A J, McMichael A, et al. A specific inhibitor of the epidermal growth factor receptor tyrosine kinase[J]. Science, 1994, 265 (5175): 1093-1095.
[3]刘刚, 李晓燕. 喹唑啉类化合物生物活性研究进展[J]. 药学进展, 2007, 31 (12): 542-550.
[4]邹立伟, 薛伟. 氨基喹唑啉类化合物抗肿瘤药物研究进展[J]. 广州化工, 2010, 38 (5): 8-10.
[5]张英, 王培义, 胡德禹, 等. 具有表皮生长因子受体抑制活性的喹唑啉衍生物研究进展[J]. 中国化学会会刊, 2012, 32: 444-461.
[6]Hynes N E, Lane H A. ERBB receptors and cancer: the complexity of targeted inhibitors [J]. Nat Rev Cancer, 2005, 5 (5): 341-354.
[7]Ban H S, Tanaka Y, Nabeyama W, et al. Enhancement of EGFR tyrosine kinase inhibition by C-C multiple bonds -containing anilinoquinazolines [J]. Bioorg Med Chem, 2010, 18 (2): 870-879.
[8]Li R D, Zhang X, Li Q Y, et al. Novel EGFR inhibitors prepared by combination of dithiocarbamic acid esters and 4-anilinoquinazolines[ J]. Bioorg Med Chem Lett, 2011, 21 (12): 3637-3640.
[9]Xu Y Y, Li S N, Yu G J, et al. Discovery of novel 4-anilinoquinazoline derivatives as potent inhibitors of epidermal growth factor receptor with antitumor activity [J]. Bioorg Med Chem, 2013, 21 (19): 6084-6091. [10]Kobayashi S, Boggon T J, Dayaram T, et al. EGFR mutation and resistance of non–small-cell lung cancer to gefitinib [J]. N Engl J Med, 2005, 352 (8): 786-792.
[11]Tsou H R, Mamuya N, Johnson B D, et al. 6-Substituted-4-(3-bromophenylamino) quinazolines as putative irreversible inhibitors of the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor (HER-2) tyrosine kinases with enhanced antitumor activity [J]. J Med Chem, 2001, 44 (17): 2719-2734.
[12]Higa G M, Abraham J. Lapatinib in the treatment of breast cancer[J]. Expert Rev Anticancer Ther, 2007, 7 (9): 1183-1192.
[13]Yap T A, Vidal L, Adam J, et al. Phase I trial of the irreversible EGFR and HER2 kinase inhibitor BIBW 2992 in patients with advanced solid tumors [J]. J Clin Oncol, 2010, 28 (25): 3965-3972.
[14]Zhang X, Li R, Qiao K, et al. Novel dithiocarbamic acid esters derived from 6-aminomethyl-4-anilinoquinazolines and 6-aminomethyl-4-anilino-3-cyanoquinolines as potent EGFR inhibitors [J]. Arch Pharm, 2013, 346 (1): 44-52.
[15]Li D D, Qin Y J, Sun J, et al. Optimization of substituted 6-salicyl-4-anilinoquinazoline derivatives as dual EGFR/HER2 tyrosine kinase inhibitors [J]. PLoS One, 2013, 8 (8): e69427.
[16]Barlaam B, Anderton J, Ballard P, et al. Discovery of AZD8931, an equipotent, reversible inhibitor of signalling by EGFR, HER2 and HER3 receptors [J]. ACS Med Chem Lett, 2013, 4 (8): 742-746.
[17]Tjulandin S, Moiseyenko V, Semiglazov V, et al. Phase I, dosefinding study of AZD8931, an inhibitor of EGFR (erbB1), HER2 (erbB2) and HER3 (erbB3) signaling, in patients with advanced solid tumors [J]. Invest New Drugs, 2013: 1-9.
[18]Ahmed M, Sadek M M, Abouzid K A, et al. In silico design, extended molecular dynamic simulations and binding energy calculations for a new series of dually acting inhibitors against EGFR and HER2 [J]. J Mol Graph Model, 2013, 44: 220-231.
[19]Ou S H. Second-generation irreversible epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs): a better mousetrap? A review of the clinical evidence [J]. Crit Rev Oncol Hematol, 2012, 83 (3): 407-421.
[20]Ferrara N. VEGF as a therapeutic target in cancer [J]. Oncology, 2005, 69 (Suppl 3): 11-16.
[21]郑军, 范松青. 靶向抗肿瘤小分子化合物类药物的研究[J]. 国际肿瘤学杂志, 2012, 39 (3): 172-175.
[22]Nakamura H, Horikoshi R, Usui T, et al. Selective inhibition of EGFR and VEGFR2 tyrosine kinases controlled by a boronic acid substituent on 4-anilinoquinazolines[ J]. Med Chem Comm, 2010, 1 (4): 282-286.
[23]Garofalo A, Goossens L, Six P, et al. Impact of aryloxy-linked quinazolines: a novel series of selective VEGFR-2 receptor tyrosine kinase inhibitors [J]. Bioorg Med Chem Lett, 2011, 21 (7): 2106-2112.
[24]Conconi M T, Marzaro G, Urbani L, et al. Quinazoline-based multi-tyrosine kinase inhibitors: synthesis, modeling, antitumor and antiangiogenic properties [J]. Eur J Med Chem, 2013, 67: 373-383.
[25]Zhang Q, Diao Y, Wang F, et al. Design and discovery of 4-anilinoquinazoline ureas as multikinase inhibitors targeting BRAF, VEGFR-2 and EGFR[J]. Med Chem Commun, 2013, 4: 979-986.
[26]楼文珠, 孙雪燕, 李龙珠, 等. 组蛋白脱乙酰酶抑制剂的抗癌作用[J]. 中国细胞生物学学报, 2011, 33 (9): 1045-1052.
[27]Cai X, Zhai H X, Wang J, et al. Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy) N-hydroxyheptanamide (CUDC-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer [J]. J Med Chem, 2010, 53 (5): 2000-2009.
[28]Thaler F, Minucci S. Next generation histone deacetylase inhibitors: the answer to the search for optimized epigenetic therapies? [J]. Expert Opin Drug Discov, 2011, 6 (4): 393-404.
[29]Mahboobi S, Sellmer A, Winkler M, et al. Novel chimeric histone deacetylase inhibitors: a series of lapatinib hybrides as potent inhibitors of epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), and histone deacetylase activity [J]. J Med Chem, 2010, 53 (24): 8546-8555.
[30]Beckers T, Mahboobi S, Sellmer A, et al. Chimerically designed HDAC-and tyrosine kinase inhibitors. A series of erlotinib hybrids as dual-selective inhibitors of EGFR, HER2 and histone deacetylases[ J]. Med Chem Comm, 2012, 3 (7): 829-835.
[31]Zhang X, Su M, Chen Y, et al. The design and synthesis of a new class of RTK/HDAC dual-targeted inhibitors [J]. Molecules, 2013, 18 (6): 6491-6503.
Research Progresses in Antitumor Activity of Multiple Target 4-Substituted Anilinoquinazoline Derivatives
GONG Feihu, SUN Liping
(Department of Medicinal Chemistry, China Pharmaceutical University, Nanjing 210009, China)
4-Substituted anilinoquinazoline derivatives are a class of compounds with high activities in quinazoline tyrosine kinase inhibitors. They can inhibit the epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR) and histone deacetylase (HDAC). These compounds have become a research hotspot of antitumor drugs. The advances in research on antitumor bioactivity of 4-substituted anilinoquinazoline derivatives with multiple targets have been reviewed in this paper.
4-substituted anilinoquinazoline;human epidermal growth factor receptor;vascular endothelial growth factor receptor; histone deacetylase; multiple target; antitumor bioactivity
R979.1
A
1001-5094 (2014) 01-0025-06
·接受日期:2013-11-05
项目资助:国家自然科学基金资助项目(No.21172265);江苏省高校“青蓝工程”项目
孙丽萍,教授;
研究方向:新药分子设计与合成研究;
Tel:025-83271414;E-mail:chslp@cpu.edu.cn
猜你喜欢
能源化工(2022年1期)2023-01-14
分子催化(2022年1期)2022-11-02
能源化工(2021年6期)2021-12-30
纺织检测与标准(2021年3期)2021-12-03
中成药(2018年12期)2018-12-29
中成药(2018年12期)2018-12-29
天然产物研究与开发(2018年1期)2018-02-02
中成药(2018年1期)2018-02-02
中南大学学报(自然科学版)(2016年2期)2017-01-19
中国塑料(2016年7期)2016-04-16
