
B-Raf激酶抑制剂及其耐药机制的研究进展
2014-03-14 09:48张帆陆涛唐伟方
药学进展 2014年1期
张帆,陆涛,唐伟方
(中国药科大学理学院,江苏 南京 211198)
B-Raf激酶抑制剂及其耐药机制的研究进展
张帆,陆涛,唐伟方*
(中国药科大学理学院,江苏 南京 211198)
Raf-MEK-ERK信号转导通路是调控细胞生长、分化和增殖最重要的通路之一。在该通路中,Raf的突变会导致肿瘤的发生,尤其是B-Raf,其在肿瘤中的突变率较高,是目前抗肿瘤药物研究的重要靶标之一。综述多种常见的B-Raf激酶抑制剂及其相关耐药机制的研究进展。
B-Raf激酶抑制剂;抗肿瘤活性;耐药机制
Raf是Raf/MEK/ERK信号通路中的重要组成部分。这一通路是促丝裂原蛋白激酶(MAPK)通路中的一种,其将胞外信号如生长因子、细胞因子和激素等传递入细胞核,引起相关基因的转录,从而调控细胞的生长、增殖、分化和凋亡[1-2]。Raf家族由3个亚型构成:A-Raf、B-Raf和C-Raf (即Raf-1)。其中,B-Raf在人类肿瘤中起着重要作用,在多种人类肿瘤中均发现了突变的B-Raf,其在黑色素瘤中的突变率为66%,甲状腺癌中为36%,卵巢癌中为20%,结直肠癌中为10%[3-4]。B-Raf绝大部分突变形式为B-RafV600E,即该酶激活片段600位的缬氨酸被谷氨酸所取代,这种突变可致酶活性提高约500倍,可持续激活下游的信号级联效应器MEK和ERK,导致这条信号通路的持续激活,对肿瘤的发生、生长增殖和侵袭转移至关重要[5],现已成为抗黑色素瘤等突变肿瘤的有效作用靶标之一。由此,众多医药公司相继开发了多种B-Raf激酶抑制剂(见表1)。根据抑制剂与激酶作用的不同方式,可将B-Raf激酶抑制剂分为Ⅰ型和Ⅱ型两类。Ⅱ型抑制剂优先与激酶的非活性构象结合,如索拉菲尼(代号:BAY43-9006,商品名:多吉美,1)、瑞戈菲尼(regorafenib,2),而Ⅰ型抑制剂在活性构象中靶向于ATP 结合位点,如威罗菲尼(代号:PLX4032/RG7204,商品名:ZelboRaf,3)、dabrafenib(代号:GSK2118436,商品名:Tafinlar,4)、PLX4720(5)和SB-590885(6)。尽管这些药物在临床上都表现出可喜的疗效,但也有许多不足,引发耐药性就是问题之一。下面将重点介绍已上市的B-Raf激酶抑制剂及其引发肿瘤耐药性的机制。
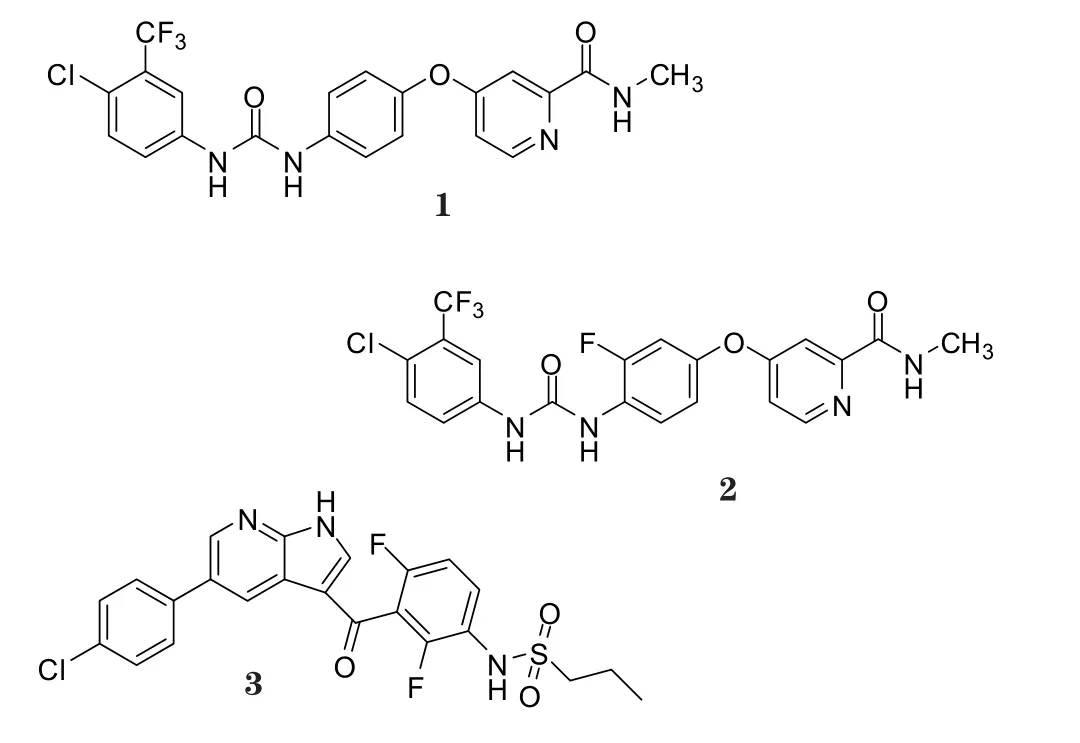


表1 B-Raf激酶抑制剂Table 1 B-Raf kinase inhibitors
1 临床常用B-Raf激酶抑制剂
1.1 索拉菲尼
索拉菲尼由拜耳公司和Onyx公司联合开发,是首个获准的口服多靶点抗肿瘤药物。本品于2005年12月获美国FDA批准用于晚期肾癌的一线治疗,于2007年11月获准用于治疗晚期肝癌,又于2013年11月获准用于甲状腺瘤。其能抑制多种激酶的活性,包括Raf激酶、VEGFR-2、VEGFR-3、PDGFR-β、KIT和FLT-3等。
作为Ⅱ型B-Raf激酶抑制剂的代表药物,索拉菲尼用于肾癌患者,总生存期仅延长2~3个月,客观缓解率(ORR)低于10%,肝癌患者使用本品后的中位总生存期(OS)也仅为10.7个月[6]。虽然索拉菲尼能适当延迟症状恶化的时间,但长时间使用可导致肿瘤产生耐药性,严重影响疗效。
1.2 威罗菲尼
威罗菲尼是由罗氏/Plexxikon制药公司开发的可用于治疗不可手术或转移性的抗黑色素瘤药物,于2011年8月17日获美国FDA批准上市,该药为一种口服有效的B-RafV600E激酶Ⅰ型强效抑制剂。在携带突变型B-RafV600E的黑色素瘤细胞中进行的研究显示,威罗菲尼可通过阻断B-RafV600E/MEK/ERK通路起到抑制细胞增殖的作用,而对携带B-RafWT的黑色素瘤细胞无效[7]。
起初,使用威罗菲尼的患者其病情有所缓解,但该药的长期疗效并不理想——大部分用药者最终疾病复发,并发展成耐药且致死的黑色素瘤。Das Thakur等[8]发现,黑色素瘤细胞对威罗菲尼产生耐药性的机制,促使肿瘤细胞对该药产生了依赖性。其结果是,黑色素瘤细胞利用威罗菲尼刺激自身快速生长,进而发展为耐药和致死性肿瘤。
1.3 Dabrafenib
Dabrafenib是由葛兰素史克公司开发的一种选择性B-RafV600E激酶Ⅰ型抑制剂,于2013年5月29日获美国FDA 批准上市,用于不可手术或转移性的黑色素瘤。研究显示,对许多患者而言,dabrafenib的疗效持续时间短,原因是肿瘤细胞产生了耐药性。目前,将dabrafenib与MEK抑制剂trametinib(代号:GSK1120212,商品名:Mekinist)联合用药的Ⅰ/Ⅱ期临床试验已经完成[9]。研究人员将162名携带B-RafV600E的黑色素瘤患者随机分为3组,分别给予dabrafenib(150 mg,bid)、dabrafenib(150 mg,bid)+trametinib(1 mg,qd),以及dabrafenib(150 mg,bid)+trametinib(2 mg,qd)。结果发现dabrafenib(150 mg,bid)+trametinib(2 mg,qd)组患者的无进展生存期明显高于dabrafenib(150 mg,bid)组患者(9.4 vs 5.8个月);在dabrafenib(150 mg,bid)+trametinib(2 mg,qd)组中,12个月肿瘤无进展的患者比例为41%,而在dabrafenib(150 mg,bid)组中这项数据仅为9%[10]。基于上述研究结果,FDA于2013年9月授予dabrafenib/trametinib联合用药的优先审查资格。
2 B-Raf抑制剂的耐药机制
随着肿瘤细胞对B-Raf激酶抑制剂耐药性的出现,近年来,研究人员努力探寻与耐药相关的具体机制,以期能找到新的治疗手段。
2.1 对Ⅰ型抑制剂产生耐药的机制
研究表明,对威罗菲尼、dabrafenib、PLX4720和SB-590885等Ⅰ型抑制剂产生耐药的机制主要包括以下2种途径:一种途径为肿瘤细胞通过COT(由MAP3K8编码)、NRAS、p61B-RafV600E、MEK等突变体重新激活ERK磷酸化[11-14],另一种为激活细胞膜表面酪氨酸激酶受体(RTK)[12,15],建立不依赖于ERK磷酸化的生存途径(见图1)。

图1 与Ⅰ型抑制剂相关的主要耐药机制Figure1 The main resistance mechanisms related to type I inhibitors
2.1.1 COT过表达 Johannessen等[11]研究发现,COT激酶过表达可激活MAPK通路,导致肿瘤细胞对PLX4720耐药。该课题组通过表达约600种激酶及其相关的开放阅读框来研究选择性B-Raf激酶抑制剂的耐药机制,最终确证,MAP3K8(编码基因为COT/Tpl2)作为MAPK通路的激活因子,可促使B-RafV600E细胞株对B-Raf抑制剂产生耐药性。COT通过依赖MEK的方式激活ERK,而不需要Raf信号。黑色素瘤复发患者使用MEK或B-Raf抑制剂后肿瘤细胞组织所产生的耐药性,以及PLX4720体外诱导B-RafV600E细胞株产生的耐药性,均与COT的过表达有关。研究表明,通过联合用药的方式(例如B-Raf/MEK抑制剂的组合或B-Raf/COT抑制剂的组合),有望抑制肿瘤耐药性的产生。
2.1.2 NRAS突变激活 Nazarian等[12]研究发现,RAS激酶亚型NRAS的突变体——NRAS(Q61K)的激活可引起肿瘤细胞对威罗菲尼的获得性耐药,而不需通过B-RafV600E的二次突变来实现。上述发现在体外耐药性诱导实验中得到了证实:肿瘤细胞经威罗菲尼处理后,产生高活性的突变体NRAS(Q61K),突变的NRAS(Q61K)再激活C-Raf,从而重新激活MAPK信号通路,导致耐药性产生。与此同时,NRAS突变的肿瘤对MEK抑制剂保持敏感性。敲除NRAS后,耐药的细胞系生长减缓。此外,在来自黑色素瘤患者、对威罗菲尼耐药的B-RafV600E阳性细胞亚系中进行的研究亦证实了NRAS突变激活在肿瘤耐药性产生过程中的重要作用。
2.1.3 B-RafV600E突变体的表达和MEK突变 Poulikakos等[13]研究发现,对威罗菲尼耐药的SKMEL-239细胞株可表达一种B-RafV600E突变体(其相对分子质量为61 000,也称p61B-RafV600E突变体),该突变体缺少外显子4—8区域(该区域包含了RAS结合结构域),从而表现出低水平的RAS激活能力。与完整的B-RafV600E相比,p61BRafV600E的二聚体形成能力增强,可重新激活ERK信号,导致细胞对B-RafV600E抑制剂耐药。在产生p61B-RafV600E的细胞内,B-Raf抑制剂无法抑制ERK信号的传导,而通过突变不再产生p61B-RafV600E的细胞,则重新恢复对威罗菲尼的敏感性。最后,Poulikakos等研究发现,19名因使用威罗菲尼而产生耐药的肿瘤患者中,有6名体内表达p61B-RafV600E。通过对相关数据进行分析,课题组确证了这种新的获得性耐药机制——p61B-RafV600E表达,并以不依赖于RAS的方式二聚化。
Wagle等[14]在1名对威罗菲尼早期应答显著、后期产生耐药的患者肿瘤样本中,前后对比发现了MEK1点突变C121S,这一突变增加了MEK1活性,导致肿瘤对B-Raf和MEK抑制剂耐药。
2.1.4 血小板衍生生长因子受体β途径 血小板衍生生长因子受体β(PDGFRβ)的过表达可引起肿瘤耐药性的产生。Nazarian等[12]在体外诱导的对威罗菲尼耐药的细胞中检测到PDGFRβ 过表达且活性增加,以及激活RTK磷酸化途径中相关酪氨酸的磷酸化。当敲除PDGFRβ后,对威罗菲尼耐药的细胞系的生长减缓。提示,PDGFRβ过表达使得对威罗菲尼敏感的肿瘤亲本细胞系产生耐药。
2.1.5 胰岛素样生长因子受体途径 Villanueva等[15]报道了一种对SB-590885耐药的黑色素瘤细胞株,该细胞株对其他选择性B-Raf抑制剂交叉耐药,其耐药机制为Raf激酶3种异构体的灵活变构。运用p-RTK芯片分析鉴定出胰岛素样生长因子受体(IGF-1R),IGF-1R不仅能通过任意一种Raf亚型激活MEK-ERK,还可以激活PI3K/AKT等与细胞增殖和凋亡密切相关的信号通路。耐药细胞中IGF-1R/PI3K信号增强,而联合使用IGF-1R/PI3K和MEK抑制剂可诱导耐药细胞死亡。在肿瘤复发病人体内可见IGF-1R和pAKT表达水平增加,进一步确证了上述耐药机制。
2.1.6 EGFR代偿性激活 对于近10%携带B-RafV600E的结直肠癌患者而言,目前暂无有效的靶向治疗方案[16]。研究人员发现,威罗菲尼虽能抑制结直肠癌细胞内的B-Raf活性,但长时间使用会引发表皮生长因子受体(EGFR)的代偿性激活,为癌细胞生长提供源源不断的能量。黑色素瘤中也可发生同样的代偿反应,但该肿瘤细胞内EGFR 水平过低,不足以抵消B-RafV600E的封闭,因而不会发生耐药。这些发现提示,携带B-RafV600E突变体的结直肠癌患者或可从靶向B-Raf和EGFR的药物的联合用药方案中获益。
2.2 对Ⅱ型抑制剂产生耐药的机制
针对索拉菲尼这种Ⅱ型抑制剂的研究表明,肿瘤细胞中存在多种旁路机制,促使其对索拉菲尼产生获得性耐药。
2.2.1 磷脂酰肌醇-3-羟激酶/蛋白激酶B通路 磷脂酰肌醇-3-羟激酶/蛋白激酶B(PI3K/Akt)和MAPK通路在肝癌组织中被激活或过度表达的比例很高。PI3K/Akt通路与MAPK通路之间存在交叉(crosstalk),两条通路上游均为RAS激酶,当索拉菲尼靶向抑制酪氨酸激酶与MAPK通路时,并行的PI3K/Akt通路并不受影响,这种潜在的代偿机制导致肿瘤对索拉菲尼耐药性的产生。Chen等[17]研究显示,在长期给药后导致对索拉菲尼耐药的肝癌细胞中,磷酸化的Akt和p85蛋白过度表达。同样,Akt异构表达的肝癌细胞也表现出对索拉菲尼的耐药性。此外,当敲除Akt基因或使用Akt抑制剂MK-2206时,肿瘤细胞对索拉菲尼的耐药现象发生逆转。
2.2.2 Janus激酶/信号转导和转录激活因子通路 Janus激酶/信号转导和转录激活因子(JAK-STAT)通路参与调控细胞增殖、分化、生存、迁移与凋亡[18-19]。STAT3在基因转录调控中起着关键的作用,可被许多JAK激酶介导的生长因子受体(如PDGFR、FGFR、EGFR)和细胞因子激活[20]。STAT3在肝癌细胞中被激活,敲除STAT3蛋白基因对肝癌细胞可起到增殖抑制作用[21]。通过抑制Src同源蛋白酪氨酸磷酸酶(SHPS,如SHP-1和SHP-2),可负调控STAT[18]。索拉菲尼以依赖SHP-1的方式对STAT3起到抑制作用[22]。研究发现,对索拉菲尼耐药的肝癌细胞能过表达p-STAT3、p-JAK1和p-JAK2,而SHP-1和p-SHP-1的表达水平较低,提示JAK-STAT信号通路参与肝癌细胞对索拉菲尼的耐药性形成过程[20]。
2.2.3 上皮细胞-间充质转化 上皮细胞-间充质转化(EMT)是指上皮细胞通过特定程序转化为具有间质表型细胞的生物学过程,该过程参与胚胎发育和伤口愈合,并对癌症的转移与侵袭产生重要作用[23-24]。EMT受上游通路(如MAPK、PI3K/Akt)调控[25]。最新研究表明,EMT参与肿瘤耐药性的产生过程,而靶向EMT可扭转肿瘤的耐药性[26]。EMT在肝癌细胞对舒尼替尼产生耐药性的过程中的作用已被报道[27]。一项在肝癌细胞中进行的研究表明,索拉菲尼可通过MAPK信号下调锌指转录因子SNAI-1的表达,从而抑制肝细胞生长因子(HGF)诱导的EMT途径[25]。上述研究表明,EMT可能参与肝癌细胞对索拉菲尼的耐药性形成过程,但具体的机制还有待进一步研究。
除了上述机制,一些研究还表明,EGFR、葡萄糖调节蛋白78(GRP78)、多药耐药蛋白2(MDRP2)、NF-κβ、自噬及缺氧微环境也可能参与肝癌组织对索拉菲尼产生获得性耐药的过程[28-33]。
3 结语
多种B-Raf激酶抑制剂已经在临床上显示出巨大的突破性疗效,大多数患者在治疗初期其肿瘤生长被很好地抑制,但部分患者在长期给药后出现耐药现象。目前已经通过临床前及部分临床数据确证了一些耐药机制。其中,修复MAPK信号通路,以及激活替代增殖生存途径,是2种最为常见的旁路机制。随着B-Raf激酶抑制剂耐药机制不断地被发现和阐明,可以相信,这将会更快更好地促进联合用药、间歇性给药以及个体化治疗等新的治疗策略的发展,同时也为多靶点抗肿瘤药物的开发提供了一定思路。
[1]Howe L R, Leevers S J, Gomez N, et al. Activation of the MAP kinase pathway by the proteinkinase Raf [J]. Cell, 1992, 71 (2): 355-342.
[2]Yang S H, Sharrocks A D, Whitmarsh A J, et al. Transcriptional regulation by the MAP kinase signaling cascades [J].Gene, 2003, 320: 3-21.
[3]Xu X L, Quiros R M, Gattuso P, et al. High prevalence of BRAF gene mutation in papillary thyroid carcinomas and thyroid tumor cell lines [J]. Cancer Res, 2003, 63 (15): 4561-4567.
[4]Fransen K, Klintenas M, Osterstrom A. Mutation analysis of the BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas [J].Carcinogenesis, 2004, 25 (4): 527-533.
[5]Wan P T C, Garnett M J, Roe S M, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF [J]. Cell, 2004, 116 (6): 855-867.
[6]张惠洁, 郭卫东. 索拉菲尼在肿瘤治疗中的研究进展[J]. 中华临床医师杂志, 2013, 7 (1): 258-260.
[7]Yang H, Higgins B, Kolinsky K, et al. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models [J]. Cancer Res, 2010, 70 (13): 5518-5527.
[8]Das Thakur M, Salangsang F, Landman A S, et al. Modelling vemuRafenib resistance in melanoma reveals a strategy to forestall drug resistance [J]. Nature, 2013, 494 (7436): 251-255.
[9]Aplin A E, Kaplan F M, Shao Y. Mechanisms of resistance to RAF inhibitors in melanoma [J]. J Invest Dermatol, 2011, 131 (9): 1817-1820.
[10]Flaherty K T, Infante J R, Daud A , et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations [J]. N Engl J Med, 2012, 367 (18): 1694-1703.
[11]Johannessen C M, Boehm J S, Kim S Y, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation [J]. Nature, 2010, 468 (7326): 968-972.
[12]Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation [J]. Nature, 2010, 468 (7326): 973-977.
[13]Poulikakos P I, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) [J]. Nature, 2011, 480 (7377): 387-390.
[14]Wagle N, Emery C, Berger M F, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling [J]. J Clin Oncol, 2011, 29 (22): 3085-3096.
[15]Villanueva J, Vultur A, Lee J T, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K [J]. Cancer Cell, 2010, 18 (6): 683-695.
[16]Corcoran R B, Ebi H, Turke A B, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemuRafenib [J]. Cancer Discov, 2012, 2 (3): 227-235.
[17]Chen K F,Chen H L, Tai W T, et al. Activation of phosphatidylinositol 3-kinase/Akt signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells [J] . J Pharmacol Exp Ther, 2011, 337 (1): 155-161.
[18]Smirnova O V, Ostroukhova T Y, Bogorad R L. JAK-STAT pathway in carcinogenesis: is it relevant to cholangiocarcinoma progression? [J]. World J Gastroenterol, 2007, 13 (48): 6478-6491.
[19]Fabregat I. Dysregulation of apoptosis in hepatocellular carcinoma cells [J]. World J Gastroenterol, 2009, 15 (5): 513.
[20]Tai W T, Cheng A L, Shiau C W, et al. Dovitinib induces apoptosis and overcomes sorafenib resistance in hepatocellular carcinoma through SHP-1-mediated inhibition of STAT3 [J]. Mol Cancer Ther, 2012, 11 (2): 452-463.
[21]Gu F M, Li Q L, Gao Q, et al. Sorafenib inhibits growth and metastasis of hepatocellular carcinoma by blocking STAT3 [J] . World J Gastroenterol, 2011, 17 (34): 3922-3932.
[22]Tai W T, Cheng A L, Shiau C W, et al. Signal transducer and activator of transcription 3 is a major kinase-independent target of sorafenib in hepatocellular carcinoma [J]. J Hepatol, 2011, 55: 1041-1048.
[23]Maheswaran T, Rushbrook S M. Epithelial-mesenchymal transition and the liver: role in hepatocellular carcinoma and liver fibrosis [J]. J Gastroenterol Hepatol, 2012, 27: 418-420.
[24]van Zijl F, Zulehner G, Petz M, et al. Epithelial-mesenchymal transition in hepatocellular carcinoma [J]. Future Oncol, 2009, 5 (8): 1169-1179.
[25]Nagai T, Arao T, Furuta K, et al. Sorafenib inhibits the hepatocyte growth factor-mediated epithelial mesenchymal transition in hepatocellular carcinoma [J]. Mol Cancer Ther, 2011, 10 (1): 169-177.
[26]Wang Z, Li Y, Ahmad A, et al. Targeting miRNAs involved in cancer stem cell and EMT regulation: an emerging concept in overcoming drug resistance [J]. Drug Resist Updat, 2010, 13 (4/5): 109-118.
[27]Marijon H, Dokmak S, Paradis V, et al. Epithelial-to-mesenchymal transition and acquired resistance to sunitinib in a patient with hepatocellular carcinoma [J]. J Hepatol, 2011, 54 (5): 1073-1078.
[28]Blivet-Van Eggelpoël M J, Chettouh H, Fartoux L, et al. Epidermal growth factor receptor and HER-3 restrict cell response to sorafenib in hepatocellular carcinoma cells [J]. J Hepatol, 2012, 57 (1): 108-115.
[29]Chiou J F, Tai C J,Huang M T, et al. Glucose-regulated protein 78 is a novel contributor to acquisition of resistance to sorafenib in hepatocellular carcinoma [J]. Ann Surg Oncol, 2010, 17 (2): 603-612.
[30]Shibayama Y, Nakano K, Maeda H, et al. Multidrug resistance protein 2 implicates anticancer drug-resistance to sorafenib [J]. Biol Pharm Bull, 2011, 34 (3): 433-435.
[31]Wu J M, Sheng H, Saxena R, et al. NF-kappaB inhibition in human hepatocellular carcinoma and its potential as adjunct to sorafenib based therapy [J]. Cancer Lett, 2009, 278 (2): 145-155.
[32]Shi Y H, Ding Z B, Zhou J, et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis [J]. Autophagy, 2011, 7 (10): 1159-1172.
[33]Bottsford-Miller J N, Coleman R L, Sood A K. Resistance and escape from antiangiogenesis therapy: clinical implications and future strategies [J]. J Clin Oncol, 2012, 30 (32): 4026-4034.
Advances in Research on B-Raf Kinase Inhibitors and Their Drug Resistance Mechanisms
ZHANG Fan, LU Tao, TANG Weifang
(School of Sciences, China Pharmaceutical University, Nanjing 211198, China)
Raf-MEK-ERK signal transduction pathway is one of the most important pathways regulating the cell growth, differentiation and proliferation. In this pathway, Raf kinase mutation can cause tumor genesis, especially the B-Raf, whose mutation rate is very high among the tumors. B-Raf has become one of the most important targets in the anti-tumor drug study. The advances in research on B-Raf kinase inhibitors and their drug resistance mechanisms were reviewed in this paper.
B-Raf kinase inhibitor; anti-tumor activity; drug resistance mechanism
R979.1
A
1001-5094 (2014) 01-0031-05
接受日期:2013-11-20
项目资助:中央高校基本科研业务费专项基金(No.JKZ2011005);*
唐伟方,副教授;
研究方向:抗肿瘤药物研究;
Tel:025-86185182;E-mail:tangwf126@126.com
猜你喜欢
昆明医科大学学报(2022年8期)2022-07-31
意林(2022年2期)2022-02-13
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
实用肿瘤学杂志(2020年6期)2020-12-09
世界知识画报·艺术视界(2020年6期)2020-07-04
西南国防医药(2016年6期)2016-12-01
中国医药生物技术(2015年4期)2015-12-26
青岛画报(2015年8期)2015-09-17
中国药理学通报(2014年2期)2014-05-09
