
毛细管气相色谱法测定牛奶中的甜蜜素
2014-02-11 02:51杨盛春张利刚何丽仙
大理大学学报 2014年8期
杨盛春,张利刚,周 萍,何丽仙
(大理学院药学与化学学院,云南大理 671000)
毛细管气相色谱法测定牛奶中的甜蜜素
杨盛春,张利刚,周 萍,何丽仙*
(大理学院药学与化学学院,云南大理 671000)
目的:研究用毛细管气相色谱法测定牛奶中甜蜜素含量的分析方法,通过对沉降剂用量、酯化时间等条件的优化,降低方法的检出限。方法:利用甜蜜素(环己基氨基磺酸钠)在硫酸介质中与亚硝酸钠反应后生成环己醇亚硝酸酯,用正己烷萃取,并用气相色谱检测环己醇亚硝酸酯的含量。结果:甜蜜素的线性范围为0.05~0.4 mg∕mL,方法检出限为2 μg,加标回收率在99.4%~102.5%之间,RSD为1.4%。结论:该方法简便、快捷、准确度高、重现性好,方法检出限低,优于GB∕T5009.97-2003法的4 μg检出限。
甜蜜素;牛奶;气相色谱法;毛细管柱
甜蜜素(环己基氨基磺酸钠)是人工合成的甜味剂,经研究结果表明甜蜜素有致癌的可能性,所以美国联邦法律禁止甜蜜素用作食品添加剂〔1〕。在GB2760—1996《食品添加剂使用卫生标准》中对甜蜜素的添加量作了明确的规定,乳饮料及蛋白饮料中添加的最高限量为0.65 g∕kg〔2〕。食品中甜蜜素的测定标准是GB∕T5009.97-2003〔3〕,该标准中测定甜蜜素的方法有气相色谱法(采用填充柱)、比色法和薄层层析法〔4〕。牛奶中的蛋白质有醇蛋白、乳白蛋白和乳球蛋白,这3种蛋白质生理价值高、消化吸收好,是优质蛋白质。牛奶因营养全面,而成为人们生活中的必需品。因此实时、快速、准确地检测牛奶制品中的添加成分和物质含量是提高乳制品质量的首要条件。在检测食品和饮料中的甜蜜素时GC∕FID已被广泛应用〔5-9〕,但由于在处理乳制品(高糖、高脂、高蛋白)的酯化过程中,易发生乳化现象,导致回收率偏低。因此本文试图通过对沉降剂用量、酯化时间的优化,减少酯化过程中的乳化现象,提高样品的回收率,降低检出限。
1 仪器与材料
1.1检测条件进样体积:1 μL;毛细管柱:ZBWAX;进样口温度:160.0℃;检测器:FID;柱温75℃;分流比:10∶1;载气:氮气;线速度:23.6 cm∕s;柱流量:0.8 mL∕min;H2流量:40 mL∕min;空气流量:400 mL∕min。
1.2实验仪器岛津GC-2010plus气相色谱;SPH-300氢气发生器、SPH-3空气发生器(上海仪电分析仪器有限公司);AL204电子天平(上海有限公司);AS20500BDJ超声波清洗器;M37610-33CN涡旋器(Thermo Scientific);LD4-2A离心机(北京医用离心机厂)。
1.3化学试剂甜蜜素标准品(Supelco公司,色谱纯);沉降剂(同体积的100 g∕L乙酸锌溶液+32 g∕L亚铁氰化钾),氯化钠、正己烷、硫酸、亚硝酸钠、无水硫酸钠等试剂均为分析纯。市售大理某品牌牛奶(出厂日期:2012年5月12日)。
2 试验方法
2.1蛋白质沉降精密称取充分震荡混合均匀后的试样20.0 g于100 mL具塞比色管中,加入10 mL蒸馏水,再加入5 mL沉降剂(卡瑞试剂),蒸馏水定容至刻度,充分混匀,静置30 min待蛋白质沉降完全后,过滤,收集续滤液备用。
2.2酯化反应待上述续滤液静置澄清后,精密移取20 mL于100 mL具塞比色管中,加入10 mL蒸馏水,加入50 g∕L亚硝酸钠溶液和100 g∕L硫酸各5 mL,充分混匀,放入冰箱,静置30 min,然后准确加入10 mL正己烷和5 g氯化钠,漩涡器上充分震荡1 min,静置,待有机相与水相分层后,离心分离,有机层经无水硫酸钠脱水,取适量于进样瓶中,进样分析,以峰面积外标法定量。
2.3对照品溶液制备精密称取甜蜜素标准品50 mg于50 mL容量瓶中,加蒸馏水定容,摇匀,即得1.0 mg∕mL的标准溶液。
3 结果与分析
3.1色谱图甜蜜素标准色谱图见图1。
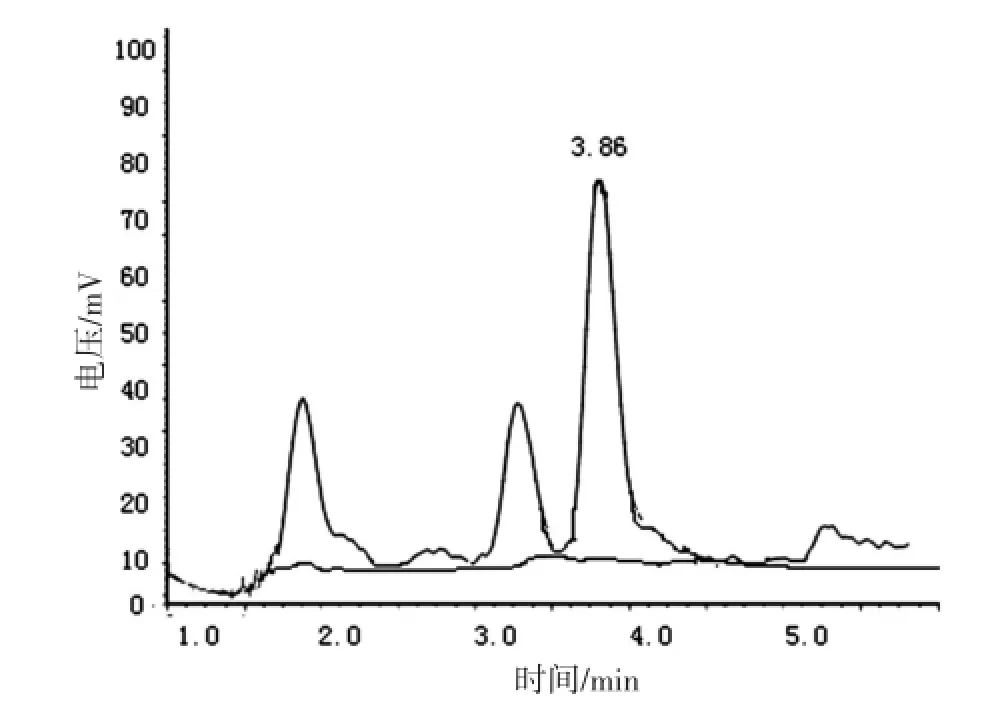
图1 甜蜜素色谱图
3.2沉降剂用量的选择分别精密称取20.0 g震荡混合均匀后的试样6份,于6支100 mL具塞比色管中,加入10 mL蒸馏水,准确移取5.0 mL甜蜜素标准品(1.0 mg∕mL)于上述样品中,分别加入卡瑞试剂:0、1、3、5、7、10 mL,蒸馏水定容至刻度,充分混均,静置30 min待蛋白质沉降完全后,过滤,收集续滤液。精密移取20 mL按“2.1.2”处理样液,并按“1.1”色谱条件进样测定。见表1。实验结果表明未加入卡瑞沉降剂的样品不能沉降蛋白质,正己烷与水相也不能分层,有机相呈乳白色无法进样测定,试验中以加入5 mL卡瑞试剂时蛋白质沉降最彻底,提取液经离心后液体清澈,酯化过程不发生乳化现象,有机相与水相分层明显,且峰面积最大。

表1 不同量卡瑞试剂对应的峰面积
3.3酯化时间优化试验还对最佳酯化反应时间进行了探讨,精称20.00 g的样品5份,于5支100 mL具塞比色管中,准确移取5.0 mL甜蜜素标准品(1.0 mg∕mL)于上述样品中,按“2.1.1”步骤处理样品,得静置澄清液,精密移取上述静置液20 mL于100 mL具塞比色管中,加入10 mL蒸馏水,加入50 g∕L亚硝酸钠溶液和100 g∕L硫酸各5 mL,充分混合均匀,放入冰箱10、20、30、40、50 min,取出后按“2.1.2”项自“准确加入10 mL正己烷和5 g氯化钠”起处理样品,并按“1.1”检测条件进行测定,以峰面积定量。见表2。

表2 酯化时间对应的峰面积
实验结果表明,起初反应剧烈,已得到大部分酯化产物,随着反应时间增加,酯化产物缓慢增加,30 min达最大值,随反应时间延长终产物反而降低。因此,本实验选酯化时间为30 min。
3.4线性范围与检出限取浓度为1 mg∕mL的对照品溶液适量,配置浓度为0.05、0.10、0.15、0.20、0.30、0.40 mg∕mL系列标准溶液,按“2.1.2”步骤处理,按“1.1”检测条件,由低到高浓度依次进样(每个浓度各进样2次,取平均值),以甜蜜素浓度(X)为横坐标,峰面积(Y)为纵坐标绘制标准工作曲线,在0.05~0.4 mg∕mL范围内线性关系良好,回归方程为Y=165 596.4X+201.65,r=0.999 4(n=6)。
当信噪比S∕N=3时,该方法的方法检出限为2 μg,高于GB∕T5009.97-2003中的4 μg,说明此方法远优于国标。
3.5精密度按“1.1”检测条件,重复测定添加5.0 mL标准溶液并按“2.1.1”、“2.1.2”处理的样品6次,得RSD为2.7%,结果表明相对标准偏差小于5%,能满足分析化学的要求。
3.6稳定性试验按“1.1”检测条件,每日测定“3.5”项处理好的样品液。见表3。结果表明:标准溶液可稳定3 d,随天数的增加,出现的杂质峰也逐渐增加。

表3稳定性试验
3.7回收率称取大理某品牌的甜牛奶(未检出甜蜜素)6份样品,按表4加入甜蜜素标准液5 mL(1 mg∕mL)。按“2.1.1”、“2.1.2”处理样品,并按“1.1”检测条件测定,计算加标回收率。由表4可知回收率在99.4%~102.5%之间。

表4 加标回收率试验结果(n=6)
4 讨论
牛奶中蛋白质的存在会加剧酯化过程中的乳化现象,使有机相与水相不易分层,而卡瑞是一种广泛使用的沉降剂,能很好的消除酯化过程中的乳化现象,且卡瑞试剂不出峰,故不影响分析测定。高温会导致酯化反应过程中生成的亚硝酸分解,不利于酯化反应的进行,故酯化反应时必须放置在0℃冰箱中。本文通过沉降剂用量、酯化时间等实验条件的优化,消除了甜蜜素测定过程中的乳化现象,提高了样品的回收率和方法的检出限,检出限优于国标法。
〔1〕苏扬,张聪.甜蜜素及检测方法〔J〕.江苏调味副食品,2011,28(1):12.
〔2〕GB2760—1996,食品添加剂使用卫生标准〔S〕.北京:中国标准出版社,2005.
〔3〕GB∕T 5009.97-2003,食品中环己基氨基磺酸钠的测定〔S〕.北京:中国标准出版社,1997.
〔4〕郑建仙.高效甜味剂和甜味抑制剂的市场现状与发展展望〔J〕.食品与机械,2006,22(1):2-3.
〔5〕管华玫,宋志敏.毛细管柱气相色谱法测定食品中甜蜜素〔J〕.中国卫生检验杂志,2009,3(19):524-525.
〔6〕梅文泉,黎其万,汪禄祥,等.毛细管柱气相色谱法测定食品中甜蜜素含量〔J〕.西南大学学报:自然科学版,2009,31(1):78-81.
〔7〕张均媚,刘伟娟,薛刚.含乳饮料中甜蜜素的测定〔J〕.食品研究与开发,2005,26(1):136-137.
〔8〕陈丽萍,邓远玲.雪糕中环己基氨基磺酸钠的气相色谱测定法〔J〕.职业与健康,2004,20(3):31-32.
〔9〕岳志坚.毛细管气相色谱法测定白酒中甜蜜素〔J〕.理化检验:化学分册,2008,44(8):785-787.
*通信作者:何丽仙,副教授.
(责任编辑 毛本勇)
The Application of Capillary Gas Chromatography in Determination of Sodium Cyclamate in Milk
YANG Shengchun,ZHANG Ligang,ZHOU Ping,HE Lixian*
(College of Pharmacy and Chemistry,Dali University,Dali,Yunnan 671000,China)
Objective:To study the analysis method of capillary gas chromatography in determination of sodium cyclamate content in milk and to lower the detection limit of the method by optimizing the settling agent dosage,esterification time and other conditions.Methods:Sodium cyclamate reacted in the medium of sulfuric acid with sodium nitrite and cyclohexanol nitrite was produced and extracted with n-hexane,and its content was determined by capillary gas chromatography.Results:Linear range was from 0.05 to 0.4 mg∕mL,the detection limit was 2 μg,recovery of standard addition was 99.4%-102.5%,and RSD was 1.4%.Conclusion:The method was simple,rapid and accurate,with good reproducibility and low detection limit,much better than detection limit of 4 μg by GB∕T5009.97-2003 method.
sodium cyclamate;milk;gas chromatography;capillary column
TS201.6
A
1672-2345(2014)08-0057-03
10.3969∕j.issn.1672-2345.2014.08.019
2013-12-25
2014-03-06
杨盛春,讲师,主要从事光谱分析、有机合成研究.
猜你喜欢
医疗卫生装备(2022年2期)2022-03-16
煤气与热力(2021年4期)2021-06-09
中国茶叶(2017年1期)2018-01-04
化学反应工程与工艺(2016年1期)2016-12-15
天然产物研究与开发(2016年11期)2016-06-15
云南中医学院学报(2015年1期)2015-07-31
现代工业经济和信息化(2015年21期)2015-02-26
西安建筑科技大学学报(自然科学版)(2014年1期)2014-11-12
天津化工(2013年3期)2013-11-05
