
碳钛复合材料负载钴基费-托合成催化剂的结构和催化性能
2013-11-26 05:45李金林张煜华
中南民族大学学报(自然科学版) 2013年4期
李金林,李 倩,张煜华
(中南民族大学催化材料科学湖北省暨国家民委-教育部共建重点实验室,武汉430074)
费-托合成催化剂常用 Al2O3、SiO2、TiO2和碳材料等为载体.TiO2载体与活性金属间有很强的金属-载体相互作用(SMSI),可有效地提高金属的分散度,但还原度降低,TiO2被部分还原成TiOx,TiOx迁移至活性金属钴的表面对钴进行包裹和修饰,使催化剂的活性降低[1].Davis等[2]发现钌助剂的加入降低了Co/TiO2催化剂的还原温度,促进了与载体有相互作用的钴物种的还原,提高了催化剂的费-托合成反应活性.Regalbuto_等[3]采用强静电吸附方法(SEA)制备了Mn/Co/TiO2催化剂,使助剂和金属紧密接触,MnO2将钴前驱体包围,在催化剂还原过程中,催化剂发生重构,MnOx从活性金属钴表面移走,钴活性位的暴露使催化剂在费-托合成反应中性能优异.但以上研究主要是通过加入不同的助剂和改变助剂的加入方法来提高催化剂的催化性能,对TiO2载体本身进行改性鲜有报道.
碳载体是一种惰性载体,与活性金属之间作用力较弱,碳的表面还可引入一些官能团,这些官能团的存在有利于钴物种的分散,防止在催化剂焙烧和还原过程中活性金属颗粒的团聚和烧结[4].由于碳表面自由能较低,固体有使表面自由能减少达到稳定的趋势,故采用碳材料对TiO2进行表面包覆改性有望抑制金属-载体相互作用,进而有效抑制TiOx迁移,使更多钴活性位暴露.
葡萄糖水热法合成的碳钛复合材料具有原料廉价易得、制备过程简单、无有毒试剂等特点,符合现代绿色化学的理念[5].本文采用葡萄糖水热法合成碳钛复合材料,以该材料为载体,以满孔浸渍法制备钴费-托合成催化剂,并采用 XRD、H2-TPR、XPS、氮气物理吸附脱附、TEM等对催化剂进行了表征,在固定床反应器中评价了催化剂的费-托合成催化性能,探讨催化剂结构和性质对催化性能的影响规律.
1 实验部分
1.1 样品、试剂和仪器
商业TiO2(P25)(Degussa,中国分公司),葡萄糖(C6H12O6,国药集团化学试剂有限公司),硝酸钴[Co(NO3)2·6H2O,国药集团化学试剂有限公司],乙醇(CH3CH2OH,国药集团化学试剂有限公司),所有试剂均为分析纯.
X-射线粉末衍射仪(XRD,Bruker advance D8型,Cu靶,小角0.5~4°,大角10~80°,扫描步长0.0167°),电感耦合等离子体发射光谱(ICP-AES,Optima 4300DV型,美国PE公司),物理化学吸附仪(Quantachrome Autosorb-1-C-MS),透射电子显微镜(TEM,FEI Tecnai G220型,200 kV,荷兰),催化剂多功能表征仪(AMI-200型,ZETON ALTAMIRA公司),X-射线光电子能谱(XPS,VG Multilab 2000型,美国Thermal Electron公司,Al Kα靶,污染碳的C1s峰结合能值284.6 eV),气相 色谱(MicroGC3000A型,6890N型和7890N型,美国安捷伦公司).
1.2 催化剂的制备
1.2.1 碳钛复合材料的制备
碳钛复合材料的合成:称取39.6 g葡萄糖和5 g P25混合于400 mL去离子水中,室温搅拌30 min,超声20 min后,转移混合液至聚四氟乙烯反应罐内,于180℃下晶化10 h,经过滤、洗涤,于100℃干燥12 h后得到碳钛复合材料,记作GT.
1.2.2 二氧化钛预处理
TiO2(P25)粉体加蒸馏水机械搅拌至糊状,于100℃下干燥12 h后直接研磨得到白色粉末,记作P25.
1.2.3 催化剂的制备
称取计量的Co(NO3)2·6H2O溶解在一定量的蒸馏水中,将所得钴盐溶液浸渍到载体上,再在旋转蒸发仪上空转30 min,随后在真空条件下程序升温干燥催化剂(旋转蒸发仪由50℃以10℃/h升至90℃保持2 h);将浸渍好的催化剂置于空气中阴干12 h,于100℃干燥12 h,在管式炉350℃N2气氛下焙烧 6 h 得终样,分别记作 Co-P25,Co-GT.
1.3 催化剂的表征
原位XRD检测:将催化剂装入原位反应池内,通入纯H2(30 mL/min),采用程序升温升至450℃保持10 h.分别在25℃,260℃,300℃,400℃,450℃对催化剂进行过程扫描.
催化剂中钴含量的测定:在电子天平上准确称取样品10 mg,溶解,转移至100 mL容量瓶中,定容后摇匀,再在ICP-AES上测试钴的含量.
载体和催化剂的孔结构参数测定:测试前,将样品置于200℃脱气6 h,去除环境中的水分等杂质,再在液氮冷却下于-200℃开始测试.Brunauer-Emmett-Teller(BET)模型计算比表面积,相对压力p/p0为0.05~0.30.由 N2在相对压力 p/p0为 0.99时吸附量计算孔体积.以 Barrett-Joyner-Halenda(BJH)模型计算平均孔径.
TEM测试:将少量样品分散于无水乙醇中,超声2 min,用毛细管吸取少量溶液蘸在表面附有碳膜的铜网上,待无水乙醇挥发后,将铜网放进透射电镜中测试.
氢气程序升温还原测试:称取0.05 g的催化剂装入U-型石英反应管中,将热电偶插入反应管中,监测和控制反应温度.催化剂先用氩气在温度为150℃下吹扫1 h,去除其中的水分和杂质,再将温度降至 50℃,通入 10%H2/Ar(30 mL/min),以10℃/min的程序升温至800℃,保持30 min,由热导池检测器(TCD)记录H2信号.
氢气程序升温脱附测试:称取约0.1 g催化剂于U-型石英反应管中,在纯H2(30 mL/min)条件下以10℃/min从室温升至450℃,保持12 h,降温至100℃.气体切换为氩气(10 mL/min)吹扫30 min,再在氩气下以10℃/min从100℃升温至450℃并保持2h进行H2程序升温脱附,以TCD记录H2信号,由脉冲方式进行TPD面积的积分校准.钴的分散度和晶粒直径具体计算方法如下.
假设H2在Co原子上的化学吸附是按Co︰H=1︰1(原子个数比)进行,则分散度(D)计算公式为:

假设金属钴为球形且位密度为14.6 atoms/nm2[6,7],
晶粒直径=6000×(活性金属密度×金属最大表面积(100%的分散度)×分散度).
1.4 催化剂的费-托合成活性测试
费-托合成反应在固定床反应器上进行.取0.5g催化剂与5g石英砂混合均匀后装在反应管里.憋压检漏后,通入纯 H2,空速为 6 L/h/g(25℃,0.1MPa),在常压下450℃保持10 h进行还原.还原完成后,在H2气氛下将温度降至100℃,再将气体切换成合成气[H2/CO=2,空速=4 L/h/g(0℃,0.1MPa)],将反应器的压力升至 1.0 MPa.采用程序升温将温度升至目标温度开始反应,分别用冷阱(-2℃)和热阱(100℃)收集产物.尾气成分由气相色谱仪在线分析后得到气相的组成.根据气相色谱分析费-托合成催化剂的活性(CTY,Cobalt time yield)、转化频率(TOF,turnover frequency)、CH4、CO2、C5以下和烃类化合物的选择性,具体公式如下:

2 结果与讨论
2.1 TEM图
载体和催化剂的TEM结果如图1所示.由图1a可见,碳钛复合材料GT中绝大部分TiO2颗粒被碳包裹,形成了以P25为核,碳为壳的核-壳结构.这说明水热合成中核-壳结构的形成遵循了LaMer模型(见图2),这与李亚栋等[8]报道的合成碳球的生长机理类似.由图2可知,合成的碳球外表面存在很多亲水性官能团,本文合成的碳钛复合材料的碳壳外表面也具有很多亲水性的官能团(-OH,-COOH,-CO等).图1c是催化剂Co-GT的TEM图,由图中可见钴物种均匀分散在碳钛复合材料中,这可能是由于碳表面含有的官能团有利于钴物种的分散,碳钛复合材料在浸渍钴之后结构未见明显变化,仍保持了P25为核,碳为壳的核-壳结构,说明这种复合材料的水热稳定性较好.

图1 载体和催化剂的TEM图Fig.1 TEM images of the carrier and catalysts
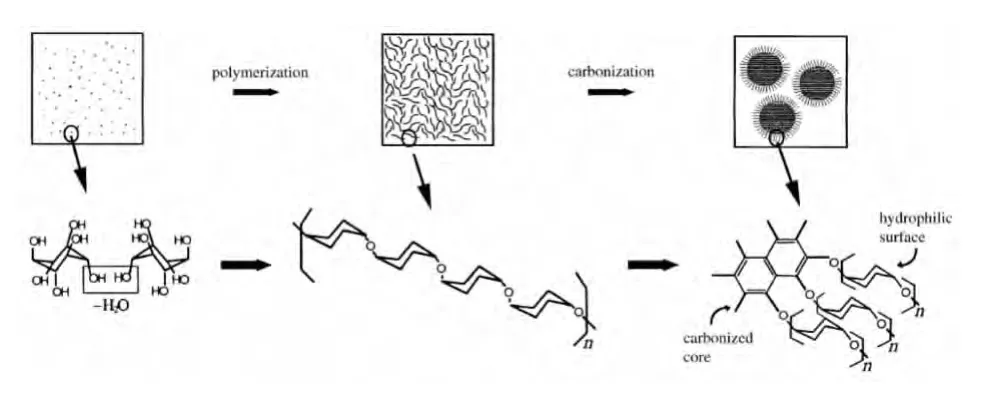
图2 LaMer模型示意图Fig.2 Schematic diagram for LaMer model
2.2 N2物理吸附脱附测试结果
催化剂Co-P25和Co-GT在H2气氛下450℃还原 10 h,还原后的催化剂记作 Co-P25-R,Co-GT-R.还原前后催化剂氮气物理吸附脱附数据如表1所示.由表1可见,碳的引入使催化剂的比表面积、孔径和孔体积均有一定程度的增加,主要是因为碳本身具有较多的微孔,比表面积的增加有利于钴物种在碳钛复合材料中的分散.微孔的出现不仅未引起平均孔径的下降反而使孔径变大,因为碳的包裹引起了孔结构的变化.由于含碳物质的气化造成Co-GT催化剂还原后的孔径、孔体积和比表面积都显著地增加[9].孔径和孔体积的增加有利于钴和反应物更好地接触,以及反应物和产物的传质、扩散.

表1 还原前后催化剂的孔结构参数Tab.1 Textural properties of the catalyst before and after reduction
2.3 电感耦合等离子体发射光谱分析结果
电感耦合等离子体发射光谱测试结果表明Co-GT-R 中钴的含量为 13.5%,Co-P25-R 中钴的含量为8.5%.对还原后的催化剂进行ICP-AES测试是由于在Co-GT催化剂还原过程中含碳物质的气化造成载体的质量损失,进而引起钴负载量增加.
2.4 XRD结果
催化剂Co-P25和Co-GT的原位X射线衍射图见图3.图3中A和R分别代表P25的两种不同晶相:A代表锐钛矿,R代表金红石.比较两图可见,碳的引入并没有引起P25晶相组成变化,TiO2晶相组成变化可引起催化剂的费-托合成催化性能的变化[10],故本文两种催化剂催化性能的差异是由于碳本身引起的,即碳的引入改变了催化剂的物理结构和化学性质,进而影响了其费-托合成催化性能.由图3a可见,Co-P25催化剂中的钴主要以Co3O4形式存在,在H2还原过程中,当还原温度达到300℃时,Co-P25催化剂中Co3O4转化为CoO,继续升温至400℃时,CoO转化为金属钴.由图3b可见,Co-GT中的钴主要以CoO形式存在,当温度升高至400℃时出现了金属钴的特征衍射峰.同样是N2气氛焙烧催化剂,Co-P25中的钴物种以Co3O4形式存在,而Co-GT中的钴主要以CoO形式存在,主要是因为催化剂中的碳的自还原作用使催化剂中的硝酸钴前驱体在N2焙烧过程中直接还原成CoO,说明在TiO2负载钴催化剂中引入碳材料有助于催化剂还原性的提高.
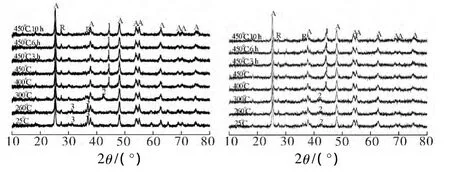
图3 催化剂的原位XRD谱图Fig.3 In situ XRD patterns of catalysts
还原后的催化剂的XRD图见图4.由图4可见,与 Co-P25-R 相比,Co-GT-R 催化剂的钴衍射峰的半峰宽更宽.由谢乐方程算得钴颗粒大小分别为:15.1 nm(Co-GT-450)和 21.2nm(Co-P25-R).这是因为碳钛复合材料的表面具有很多官能团,而这些官能团有利于钴物种的分散,与 TEM表征结果一致.

图4 催化剂 Co-GT-R 和 Co-P25-R 的 XRD 谱图Fig.4 XRD patterns of catalyst Co-GT-R and Co-P25-R
2.5 H2程序升温还原表征结果
催化剂的程序升温还原谱图见图5.对于Co-P25,305℃的还原峰归属于Co3O4→CoO的还原,369℃、437℃的峰归属于与载体相互作用程度不同的CoO→Co的还原,这与原位XRD表征结果相一致.对于Co-GT,411℃的峰归属于体相钴氧化物的还原[11],629 ℃的峰归属于含碳物质的甲烷化[12].由原位XRD结果可知,Co-GT中的钴主要以CoO存在,故体相钴氧化物的还原主要指CoO的还原.与Co-P25相比,Co-GT还原峰位置往低温方向偏移,说明Co-GT中钴还原性更好,因为碳的引入减弱了金属-载体相互作用.

图5 催化剂的H2-TPR图Fig.5 H2-TPR profiles of the catalyst
2.6 H2程序升温脱附结果
催化剂H2程序升温脱附数据见表2.由表2可见,Co-GT 催化剂分散度(52.4%)高于 Co-P25 催化剂的分散度(37.8%),说明钴在碳钛复合材料中分散得更好,这与TEM和XRD表征结果相一致.其原因主要有:1)碳的引入引起载体的比表面积和孔结构变化,比表面积的增大有利于钴更好的分散;2)碳的表面具有很多亲水性的官能团,这些官能团的存在有利于钴的分散.

表2 催化剂的H2程序升温脱附数据Tab.2 H2-temperature programmed desorption results for catalysts
2.7 X-射线光电子能谱表征结果
催化剂中Co2p电子结合能的谱图图6.由图6可见,在Co-GT催化剂的表面,钴主要以Co3O4和CoO两种形式存在[13],但在XRD图中只有CoO的特征衍射峰,由于Co3O4在催化剂体相中的相对含量较少,XRD不能检测到;但在Co-P25催化剂的表面钴主要以Co3O4物相形式存在.此外,在Co-GT催化剂中,Co3O4的 Co2p3/2电子结合能的位置为781.0 eV;在 Co-P25 催化剂中,Co3O4的 Co2p3/2电子结合能的位置为781.5 eV.与 Co-P25催化剂相比,Co-GT催化剂中Co2p3/2电子结合能位置往低能位方向偏移,表明钴物种与碳钛复合材料之间的相互作用减弱,故还原性更好,TPR表征结果也证实了这一点.

图6 催化剂的XPS谱图Fig.6 XPS spectra of the catalyst
2.8 费-托合成催化性能评价
催化剂的费-托合成活性和选择性数据见表3.表3中所列数据是从反应10 h开始反应100 h的平均数据.Co-GT 催化剂的活性(CTY)是 5.2 ×10-5mol CO/g/s,明显高于 Co-P25 催化剂的活性(2.5 ×10-5mol CO/g/s),主要因为碳钛复合材料中碳的引入抑制了TiOx的迁移,碳表面自由能较低,固体有使其表面自由能减少而稳定存在的趋势,在催化剂的还原过程中TiOx倾向于迁移至碳表面而非钴的表面,同时碳钛复合材料表面的官能团有利于钴的分散使催化剂的活性数目增加,费-托合成催化活性提高.此外,两种催化剂的TOF均在10-3数量级,说明费-托合成是一个结构非敏感性反应.与Co-P25催化剂相比,Co-GT催化剂的长链烃的选择性增强同时甲烷的选择性降低,C2~C4的选择性降低.同时由图7可见,两个催化剂在100 h反应后均明显的失活.
两种催化剂的钴的活性位的TOF基本一致,故引起两种催化剂活性(CTY)差异的主要原因是活性位数目.影响活性位数目的两个重要的因素是催化剂的分散度和还原度,碳的引入使催化剂的比表面积增大,还引入一些官能化的基团,均有利于提高催化剂的分散度;由于碳是一种惰性材料,用它包裹修饰TiO2可减弱金属-载体相互作用,促进钴物种的还原;以上这些有利因素均可引起钴活性位数目增加,促进催化剂的催化活性提高.从表3还可见,催化剂活性位数目的增加还有利于α-烯烃的重吸附,使产物重质烃选择性增加.

表3 催化剂的费-托合成反应结果Tab.3 FTS activity and selectivity of catalysts
3 结语

图7 催化剂的催化活性随反应时间的变化Fig.7 Catalytic activity with time on-stream
本文采用葡萄糖水热法合成了碳钛复合材料(GT),以该材料为载体成功制备了钴基费-托合成催化剂.与商业TiO2(P25)负载的钴催化剂(Co-P25)相比,Co-GT催化剂在费-托合成反应中表现出更优良的催化性能.原因在于:1)碳的引入使催化剂的比表面积增大,在葡萄糖水热过程中碳表面形成了较多官能团,有利于钴物种的分散,在焙烧和还原过程中,碳的微孔有利于固定这些小尺寸的钴纳米颗粒,使其不容易团聚和烧结;2)采用碳这种惰性材料对TiO2进行包裹修饰可减弱金属-载体相互作用,提高钴氧化物的还原性;3)碳材料在P25外面的包裹减弱了TiOx的迁移,这样有利于更多钴活性位的暴露,提高催化剂的催化活性.此外催化剂中活性位数目的增加有利于α-烯烃的重吸附,增加重质烃选择性.
[1]O'Shea V A,Galván M C,Prats A E,et al.Direct evidence of the SMSI decoration effect:the case of Co/TiO2catalyst[J].Chem Commun,2011,47(25):7131-7133.
[2]Jacobs G,Patterson P M,Zhang Y,et al.Fischer–Tropsch synthesis:deactivation of noble metal-promoted Co/Al2O3catalysts[J].Appl Catal A:Gen,2002,233(1/2):215-226.
[3]Feltes T E,Espinosa-Alonso L,de Smit E,et al.Selective adsorption ofmanganese onto cobaltfor optimized Mn/Co/TiO2Fischer-Tropsch catalysts[J].J Catal,2010,270(1):95-02.
[4]Sun B,Jiang Z,Fang D,et al.One-ot approach to a highly robust iron oxide/reduced graphene oxide nanocatalyst for Fischer Tropsch Synthesis[J].Chem Cat Chem,2013,5(3):714-719.
[5]Parayil S K,Kibombo H S,Wu C-M,et al.Enhanced photocatalytic water splitting activity of carbon-modified TiO2composite materials synthesized by a green synthetic approach[J].Int J of Hydrogen Ener,2012,37(10):8257-8267.
[6]Schanke D,Vada S,Blekkan E A,et al.Study of Ptromoted cobalt CO hydrogenation catalysts[J].J Catal,1995,156(1):85-95.
[7]Reuel R C,Bartholomew C H,The stoichiometries of H2and CO adsorptions on cobalt:effects of support and preparation[J].J Catal,1984,85(1):63-67.
[8]Sun X,Li Y.Colloidal carbon spheres and their Core/Shell structures with noble-metal nanoparticles[J].Angew Chem,2004,116(5):607-611.
[9]Yu G,Sun B,Pei Y,Xie S H,et al.FexOy@C spheres as an excellent catalyst for Fischer Tropsch synthesis[J].J Am Chem Soc,2009,132(3):935-937.
[10]Jongsomjit B,Wongsalee T,Praserthdam P.Characteristics and catalytic properties of Co/TiO2for various rutile:anatase ratios[J].Catal Commun,2005,6(11):705-710.
[11]Xiong H F,Zhang Y H,Wang S G,et al.Fischer–Tropsch synthesis:the effect of Al2O3porosity on the performance of Co/Al2O3catalyst[J].Catal Commun,2005,6(8):512-516.
[12]Xiong K,Liew K Y,Li J L,et al.Preparation and characterization of stable Ru nanoparticles embedded on the ordered mesoporous carbon material for applications in Fischer-ropsch synthesis[J].Appl Catal A:Gen,2010,389(1/2):173-178.
[13]Riva R,Miessner H,Vitali R,et al.Metal-support interaction in Co/SiO2and Co/TiO2[J].Appl Catal A:Gen,2000,196(1):111-123.
猜你喜欢
中学生数理化·高一版(2022年4期)2022-05-09
小学生学习指导(高年级)(2022年3期)2022-03-29
轮胎工业(2021年9期)2021-07-20
热力透平(2020年2期)2020-06-22
中学生数理化(高中版.高考数学)(2020年2期)2020-04-21
小学生学习指导(高年级)(2019年4期)2019-11-27
小学生导刊(高年级)(2017年2期)2017-06-10
中学化学(2016年5期)2016-06-04
小学生导刊(高年级)(2016年1期)2016-01-29
高中生学习·高二版(2015年3期)2015-05-21
