
蛋白质氨基酸序列的粒度概念及其在蛋白质预测中的应用
2013-09-20 09:08刘智新杨洪强包丽华
Biophysics Reports 2013年3期
刘智新, 杨洪强, 包丽华
1.山东农业大学信息科学与工程学院,应用物理系,泰安 271018;
2.山东农业大学作物生物学国家重点实验室,园艺科学与工程学院,泰安 271018;
3.山东农业大学化学与材料科学学院,泰安 271018
引 言
蛋白质氨基酸序列对蛋白质立体空间结构的形成及蛋白质功能的实现起着关键的作用,所以,蛋白质氨基酸序列的分析及其应用是蛋白质研究的重要内容之一。目前,虽然实验技术是蛋白质研究领域的主要手段,但是,蛋白质序列分析的手段在蛋白质研究、蛋白质组学研究及系统生物学研究中仍起着重要的作用。这是因为:1)在后基因组时代,产生了大量生物学数据,数据之间的关系复杂,仅通过生物学实验手段来完成这些研究任务是不现实的;2)生物学实验对具体实验环境、实验条件依赖性相对要强一些,而且做实验要消耗较多的时间,往往还需要大量的经费支持,因而制约了一些大规模研究的开展;3)虽然蛋白质氨基酸序列的研究与分析已开展了较长时间,许多技术也已经比较成熟,且在生命科学研究与实践中得到了广泛应用,但是,依然不能满足科研与实践的需求,而且,蛋白质氨基酸序列的研究与分析属于生物学基础研究领域,在这方面进行一些探索可能会产生新的生物理论突破点。
蛋白质序列分析与应用研究在技术层面主要从两个方向展开,一个是以氨基酸序列本身的排列为中心,探索蛋白质序列之间的相似性、同源性;一个是以蛋白质的氨基酸组成及各级结构为中心,探索蛋白质的生物学功能。当然,这两个方向有时是难以截然分开的。在第二个方向上,一般与蛋白质的各种预测结合得比较紧密。例如,蛋白质的二级结构类预测[1]、酶家族分类预测[2]、蛋白质的折叠速率预测[3]、蛋白质的亚细胞定位预测[4]、蛋白质的亚叶绿体定位预测[5]、凋亡蛋白定位预测[6]等。本文从蛋白质氨基酸序列的组成出发,借鉴物理学中粒度的思想,提出了蛋白质氨基酸序列的粒度概念,使用蛋白粒度对氨基酸序列进行分析,进一步给出了蛋白粒度的阶、蛋白粒度的界、蛋白粒度的极限、蛋白粒度增量等概念,得到了一些有益的结论。然后,把蛋白粒度的概念和知识应用到蛋白质二级结构类预测和凋亡蛋白的亚细胞定位预测中,取得了很好的结果。
蛋白质序列和蛋白粒度
蛋白粒度的概念
设集合B={A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y},其中,A表示丙氨酸、C表示半胱氨酸、…、Y表示酪氨酸,即集合B由20种氨基酸组成。设集合Z={z1,z2,…,zm}(1≤m≤20),且Z是B的一个子集。令集合X={x1,x2,…,xn},n是正整数。如果集合X和Z中的元素是有序的,且满足x1<x2<…<xn和z1<z2<…<zm,那么,映射f:X→Z对应一个n字母排列:f(x1)f(x2)…f(xn),且f(xi)∈Z。这个排列被称为一个字,它由从集合Z中随机选择的n个字母组成。当f(x1)≤f(x2)≤…≤f(xn)时,称f(x1)f(x2)…f(xn)为一个蛋白粒度 (protein granularity)。
蛋白粒度的阶
在上面的集合X={x1,x2,…,xn}中,n称为蛋白粒度的阶 (order)。如果在集合X中有一个元素,则称蛋白粒度的阶为1阶;如果在集合X中有两个元素,则称蛋白粒度的阶为2阶;如果在集合X中有三个元素,则称蛋白粒度的阶为3阶,依次类推。
对于一个蛋白质序列,在n阶水平上可以得到蛋白粒度的总类型数,同时,在同阶水平上,还可以得到具体一个蛋白粒度出现的频次。为了具体说明,以一蛋白质结构域序列(PDB:1RDH_A)的片段为例,这是HIV酶的一部分,由PFHGYQLEKEP这11个氨基酸组成。各阶蛋白粒度的具体提取结果见表1。
以2阶粒度提取过程为例进行说明,从序列开头首先得到第一个2阶粒度FP,然后得到第二个2阶粒度FH、第三个2阶粒度GH,如此进行下去,直到得到最后一个2阶粒度EP。其中,EK出现两次,所以,二阶粒度的总类型数等于9,FP的频次为1,EK的频次为2。

表1 蛋白结构域(PDB:1RDH_A)片段的粒度提取结果Table 1 The granularity extraction results of the protein domain sequence(PDB:1RDH_A)fragment
从粒度提取过程可以看出,蛋白粒度包含了氨基酸在序列中的排列信息,也包含了蛋白质序列的氨基酸组成信息。从表1中可以发现一个有趣的现象,10阶粒度EEFGHKLPQY的频次是2(对于序列片段“PFHGYQLEKEP”,取前10个字母,以字母表字母顺序进行排列,得到“EEFGHKLPQY”;再从第2个字母开始取后10个字母,以字母表字母顺序进行排列,得到“EEFGHKLPQY”,可以看出这两个蛋白粒度是相同的,所以,这个蛋白粒度的频次是2),这是因为第二个脯氨酸 (P)与第一个脯氨酸 (P)隔9个氨基酸相邻,所以,可以推出这种蛋白粒度反映同种氨基酸的互邻信息。
那么,一般情况下,一条蛋白质氨基酸序列的粒度分布 (频次分布亦可称为粒度谱)情况是怎样的呢?我们以CAS1A_XENLA(Swiss-Prot:P55865)这一蛋白序列为例。CAS1A_XENLA由386个氨基酸 (本文在不引起混淆的情况下,为简便起见,使用氨基酸称呼对应的氨基酸残基)组成。沿序列分别提取2阶、3阶粒度,结果见图1和图2。
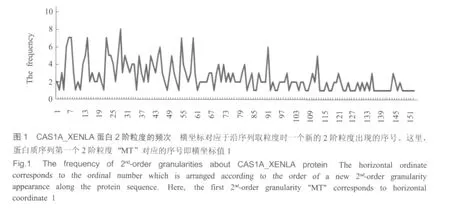
从图1可以看出,2阶粒度的频次变化范围在1到8之间,显然粒度并不是被均匀使用的,蛋白质序列对2阶粒度使用具有偏好性,但是,没有一个2阶粒度的频次处于绝对的优势地位。从图2可以看出,3阶粒度的频次变化范围在1到4之间,蛋白质序列对3阶粒度的使用也具有一定的偏好性,但也没有一个3阶粒度的频次处于绝对的优势地位。同2阶粒度相比,3阶粒度的频次最大值变小了,频次为1的粒度增多了,蛋白粒度类型的总数从153变到309,这说明蛋白质更倾向于选择不同的粒度来构成蛋白质序列,而不是靠粒度频次的变化来构成蛋白质序列。蛋白粒度类型的增加意味着蛋白质序列所携带的信息量的增大,这与蛋白质序列变长更容易形成复杂的高级空间结构的趋势相吻合。
把CAS1A_XENLA从中间分成等长的两段,然后考察每段的2阶粒度情况。为了方便比对,把每段的粒度按字母表的顺序重新排列,结果见图3(为方便比对,第二段粒度的频次被统一加上6)。从图3可以看出,两段的最大粒度频次值都是5,粒度的离散程度基本相似,有些粒度只在一段出现。计算发现第一段的2阶粒度类型数是105,第二段的2阶粒度类型数是120,这说明2阶粒度的分布在整条蛋白质序列上是不均匀、不对称的,这些性质同时为蛋白质预测提供了有益的信息。
蛋白粒度的界
定理1:给出一个n元有序集合X={x1,x2,…,xn}(x1<x2<…<xn),n是正整数。同时给出20元有序集合Z={A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y},集合Z中的20个元素即20种氨基酸。对应粒度的映射为f:X→Z,则n阶粒度水平的总粒度类型数等于从20个氨基酸中可重复选取n个氨基酸的组合数,并且这个数目为,这里,表示n阶粒度所形成的集合的元素数目,是组合数 (证明见参考文献[7])。

蛋白粒度增量
给定一个蛋白质序列,在n阶粒度水平下进一步可以得到n阶粒度类型数与n阶粒度上界的比率,则称这个比率为在n阶粒度水平下的蛋白粒度增量 (granularity increment),即

蛋白粒度的极限
对于一个具体的蛋白质序列,在不同阶粒度水平下可以得到一系列粒度类型总数,进一步研究发现,这些数目会有一个最大值,称这个最大值为这条序列蛋白粒度的极限(granularity limit)。下面用一个例子进行说明。POLG_HCVEV(Swiss-Prot:O39928)由3014个氨基酸组成 (选一个长蛋白序列是为了把情况说明得更清楚一些)。计算结果表明POLG_HCVEV的蛋白粒度极限是2807,此时对应的粒度的阶是15(见图4中的×,为方便比对,图4中的第一个数据点是该蛋白序列的长度)。从图4可以看出,随着粒度阶的增大,对应的粒度总数迅速增大,然后到达蛋白粒度的极限2807,之后缓慢变小,变小的原因是随着粒度阶的增大整条蛋白序列相对变短。2阶粒度的点是整条曲线的最低点,值为210,说明该蛋白序列已经使用了所有的2阶蛋白粒度。
当蛋白质序列的长度发生变化时,蛋白粒度类型数是如何变化的呢?下面通过新选的四条不同长度的蛋白质序列,并结合上面给出的POLG_HCVEV蛋白序列一起来进行说明。这四条新选的蛋白质序列是:CASP6_HUMAN(Swiss-Prot:P55212)、RN216_HUMAN(Swiss-Prot:Q9NWF9)、DCC_MOUSE(Swiss-Prot:P70211)和 DIDO1_MOUSE(Swiss-Prot:Q8C9B9)。CASP6_HUMAN由293个氨基酸组成,RN216_HUMAN由866个氨基酸组成,DCC_MOUSE由1447个氨基酸组成,DIDO1_MOUSE由2256个氨基酸组成,比对结果见图5(为方便比对,图5中的第一个数据点是该蛋白序列的长度)。从图5可以看出,5条蛋白序列在2阶粒度达到最低点,值分别为143、187、199、198和210,CASP6_HUMAN序列使用了143个2阶粒度,POLG_HCVEV序列使用了所有的2阶蛋白粒度。5条曲线的极值点分别为 276(4th-order)、800(6th-order)、1350(9th-order)、2052(15th-order)和 2807(15th-order)(见图5中“×”点)。
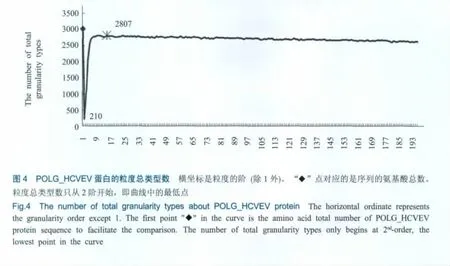
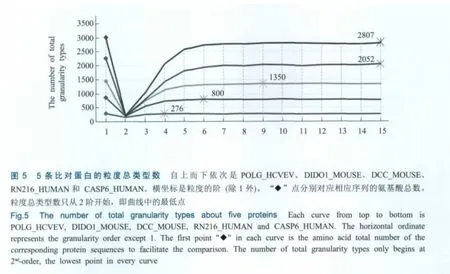
图5还显示,随着粒度阶的增加,曲线上升的总体趋势随蛋白质序列长度的增加而变得缓慢,但是在同阶粒度水平下,长序列的粒度类型数要大于短序列的粒度类型数。总体来看,随着蛋白质序列的变长,蛋白粒度的极限在更高的粒度阶水平才能达到。同时也可以得出,在同阶水平下,长序列的蛋白粒度增量要大于短序列的蛋白粒度增量,这说明粒度增量与蛋白质序列长度呈正相关效应。
利用蛋白粒度知识构建蛋白质序列特征向量
由于蛋白粒度等有关概念和知识能够反映蛋白质序列的多种组成信息,所以,可以应用蛋白粒度的有关知识对蛋白质序列进行特征抽提,抽提的特征向量在蛋白质预测中具有多种应用。下面给出一种构建蛋白质序列特征向量的具体方法。
构建蛋白质序列特征向量
对于一个蛋白质预测数据集,第k条蛋白ψ为第s类的特征向量可以表示为
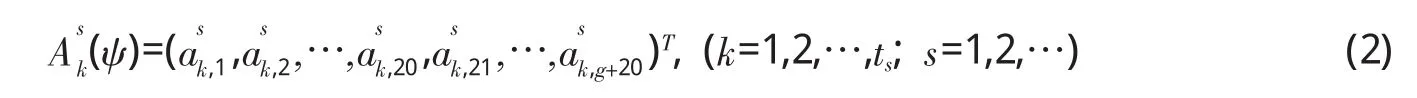
从上面的分析可以得到,蛋白粒度在一条蛋白质序列的不同片段有不同的分布,所以,可以把整条蛋白序列分成多个等长的片段以增加向量的信息含量。用表示在 n 阶粒度水平下的粒度类型数,具体的粒度特征提取方法和各符号含义见图6。
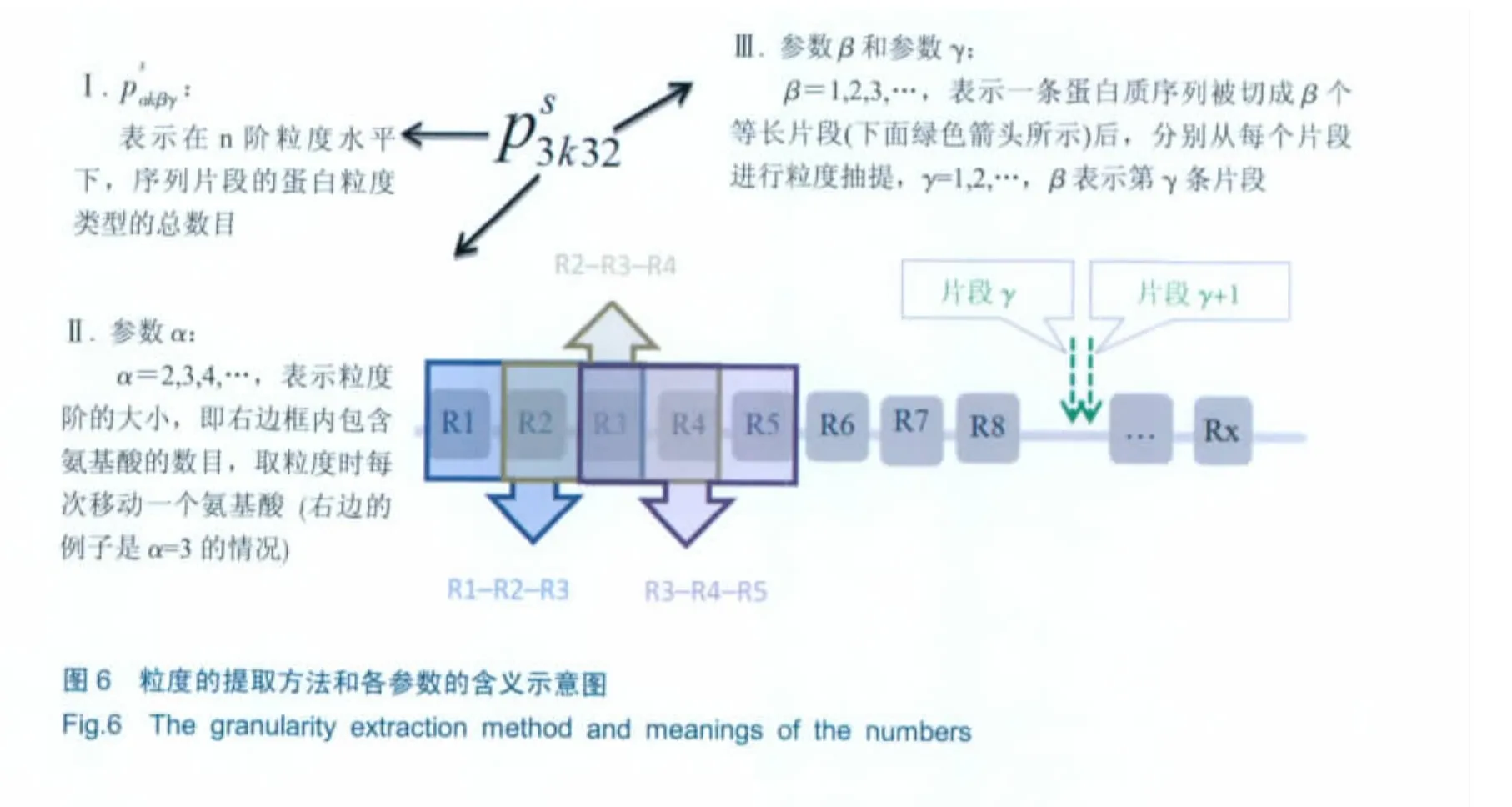
假设分别取2阶、3阶、4阶粒度,同时对整条蛋白质序列等长切割0次、1次和2次,则等式(2)中的g=18。按定理1中的结论,有

这样,最终获得了一种代表蛋白质氨基酸序列的38维特征向量
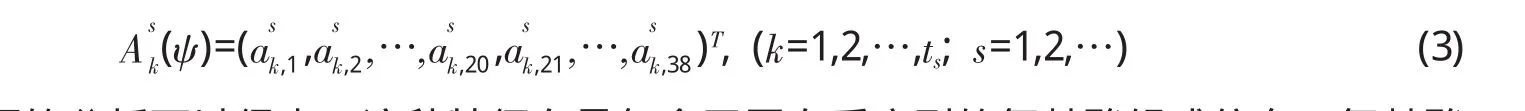
从前面的分析可以得出,这种特征向量包含了蛋白质序列的氨基酸组成信息、氨基酸排列信息、氨基酸的互邻信息、序列长度信息、蛋白粒度沿序列不对称分布等信息,所以,它能很好地代表蛋白质序列。同时,我们知道蛋白质序列的2肽表示向量是400维,3肽表示向量是8000维,4肽表示向量是160 000维,而2阶粒度表示向量是210维,3阶粒度表示向量是1540维,4阶粒度表示向量是8855维,显然,如果用粒度表示向量比用多肽表示向量具有明显的降维作用,这也是用粒度方式代表蛋白质序列所独有的优势。我们进一步推测,这种新型的表示向量在蛋白质序列同源性的高精度区分,以及由于样品不足所形成的小样品高精度区分上,将具有独特优势,而这恰恰是基因本体 (gene ontology)和蛋白质功能域 (protein function domain)等特征提取方法还略显不足的地方。
权重因子
考虑到实际蛋白质预测的需要,可以对粒度向量的各个权重因子进行进一步的优化,而不是赋予等值的权重,例如在等式(3)中,是2阶粒度增量,是3阶粒度增量,是4阶粒度增量,那么对应的权重因子可以分别设为λ1、λ2和λ3,则等式(3)变换为被赋予权重的新的38维特征向量,如(4)式。
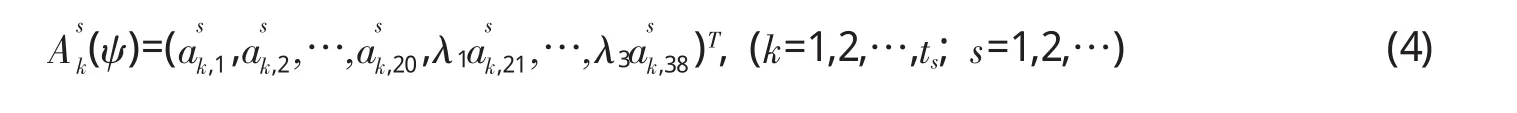
利用蛋白粒度对蛋白质二级结构类进行预测
材料和方法
标准数据集与特征提取
为了验证粒度向量对同源蛋白具有高的区分性能的推测,我们选择了Chou的蛋白质二级结构类359标准数据集[8],简写为C359集。C359集是高同源 (高于95%)蛋白质 (域)数据集,包含82个全α类蛋白质、85个全β类蛋白质、99个α/β类蛋白质和93个α+β类蛋白质。其蛋白质序列的特征提取方法见等式(4)。预测算法与评价方法
预测算法采用基于统计学习理论的支持向量机 (support vector machine,SVM)[9]。SVM被广泛用于生物数据的分析,例如基因表达数据分析[10]、蛋白质折叠识别[11]、凋亡蛋白亚细胞定位[12,13]、蛋白质分类[14]等。本实验中,支持向量机具体程序用LIBSVM[15];同时,由于SVM是二分类算法,我们采用一对一 (one-versus-one)方式来实现本实验的四分类预测。通过蛋白粒度增量和支持向量机相结合 (protein granularity increment and SVM,PGI-SVM)的方式,从而完成了蛋白质二级结构类的预测。
评价方法采用蛋白质预测中常用的Jackknife检验方法,它也是最严格、最客观的评价检验方法之一[16]。敏感系数 (Sn)、特异系数 (Sp)、Matthew相关系数 (MCC)、总体预测精度(Ac)采用蛋白质预测通用定义式[5,17]:
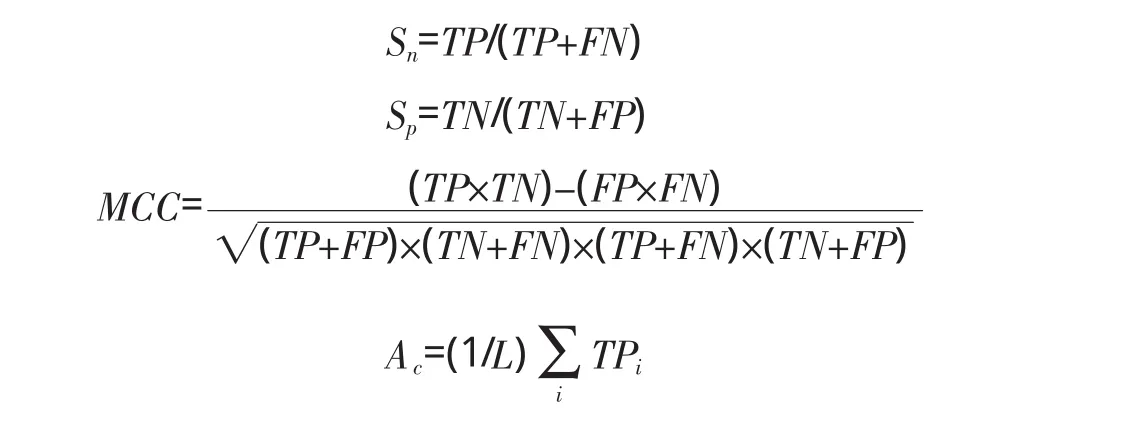
其中,TP表示真阳性数目,TN表示真阴性数目,FP表示假阳性数目,FN表示假阴性数目,L表示样品总数,i表示第i类样品。
结果和讨论
采用等式(4)构造特征向量,经过计算分析,发现当权重因子λ1=0.02、λ2=0.5、λ3=9时效果较好。对于支持向量机,采用径向基函数,然后,使用网格寻优的方式来确定最优的惩罚因子C和径向基函数的系数γ,当=1.25、log2γ=6.62时,PGI-SVM的总体预测精度 (Ac)达到了97.2%,各子集的敏感系数 (Sn)、特异系数 (Sp)、Matthew相关系数 (MCC)均达到了很高的百分比,见表2。
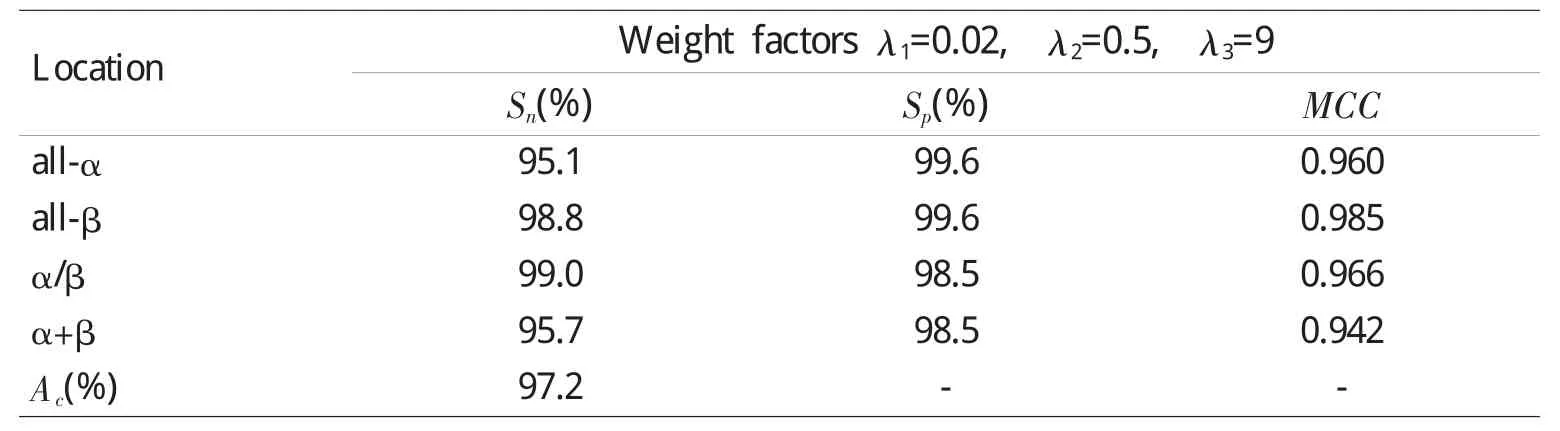
表2 PGI-SVM在C359蛋白结构类数据集的Jackknife检验结果Table 2 PGI-SVM results by the Jackknife test in the C359 protein structure class dataset
把PGI-SVM的结果与C359集Chou等的最初预测结果比对,各子集和总集的Jackknife检验精度结果列于表3。结果发现,在各个子集和总集上的预测精度都有大幅度提高,总体预测精度超过最初预测精度的13.1%。这说明粒度增量的方法在高同源的蛋白质二级结构类的预测上能够达到高精度区分的效果。

表3 不同方法在C359蛋白结构类数据集的Jackknife检验结果比对Table 3 Result comparisons of different methods by the Jackknife test in the C359 protein structure class dataset
利用蛋白粒度对凋亡蛋白定位进行预测
材料和方法
标准数据集与特征提取
为了验证粒度向量在由于样品不足所形成的小样品蛋白数据集上的高精度区分能力,我们选择了Zhou和Doctor建立的包含98条蛋白质序列的凋亡蛋白标准数据集[18],简写为ZD98集。ZD98集是凋亡蛋白集,这类功能蛋白集由于总体蛋白数目不多或新发现的蛋白序列较少而形成小的数据集,这类小数据集往往由于GO(gene ontology)条目的不完善及功能域难以确定,用基于GO等预测方法有时不如基于蛋白质序列的方法。
ZD98集包含43条细胞质蛋白 (cytoplasmic protein)、30条膜蛋白 (plasma membrane-bound protein)、13条线粒体蛋白 (mitochondrial protein)和12条其它类蛋白(other protein)。蛋白质序列的特征提取方法见等式(4)。
预测算法与评价方法
为了体现蛋白粒度特征向量具有普适性,预测算法使用K-近邻 (KNN)算法代替上面的SVM算法。K-近邻算法已经被用于各种蛋白质预测,例如:预测酶的亚家族结构类[19]、预测蛋白的亚核定位[20]、预测蛋白的亚叶绿体定位[5]等。
这里所用的是蛋白粒度增量与K-近邻算法相结合 (protein granularity increment and KNN,PGI-KNN)的预测方法。
评价方法采用蛋白质预测中常用的Jackknife检验方法,它也是最严格的评价检验方法之一[16]。敏感系数 (Sn)、特异系数 (Sp)、Matthew相关系数 (MCC)、总体预测精度 (Ac)采用蛋白质预测通用定义式 (同上)。
结果和讨论
采用等式(4)构造特征向量,经计算分析发现,当权重因子λ1=0.028571、λ2=0.066667、λ3=0.090909时效果较好。对于K-近邻算法,采用Cityblock距离函数,当K=2时,PGI-KNN的总体预测精度 (Ac)达到94.9%,各子集的敏感系数 (Sn)、特异系数 (Sp)、Matthew相关系数 (MCC)均达到了很高的百分比,见表4。
把PGI-KNN的结果与Zhou和Doctor的最初预测结果比对,各子集和总集的Jackknife检验精度结果列于表5。结果发现,在各个子集和总集上的预测精度都有大幅度提高,总体预测精度超过了最初预测精度的22.4%。这说明粒度增量的方法在这种小的蛋白功能集上能够达到高精度区分的效果。
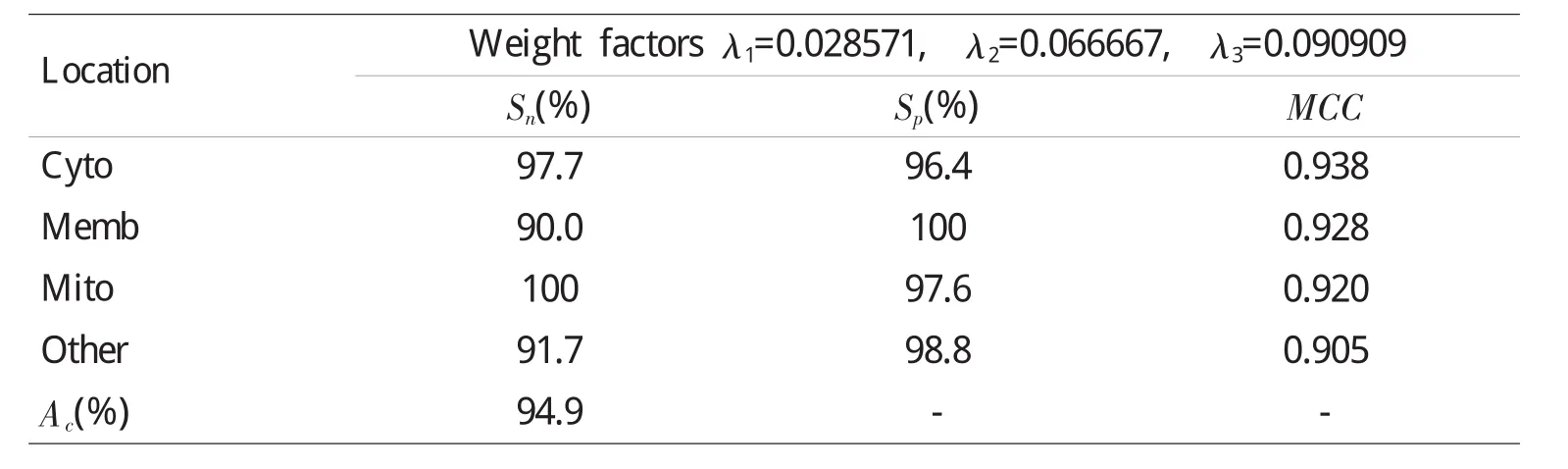
表4 PGI-KNN在ZD98凋亡蛋白数据集的Jackknife检验结果Table 4 PGI-KNN results by the Jackknife test in the ZD98 apoptosis protein dataset
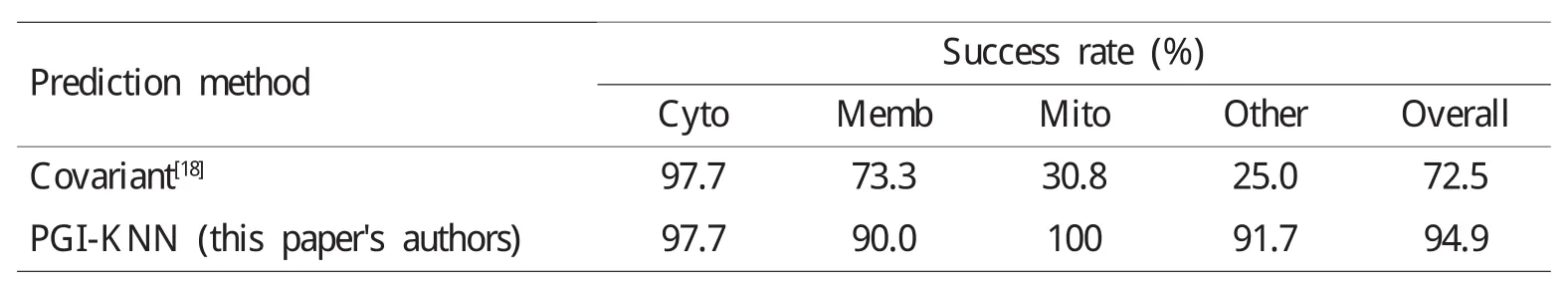
表5 不同方法在ZD98凋亡蛋白数据集的Jackknife检验结果比对Table 5 Result comparisons of the different methods by the Jackknife test in the ZD98 apoptosis protein dataset
结 论
从蛋白质氨基酸序列的组成出发,借鉴物理学中粒度的思想,提出了蛋白质氨基酸序列的粒度概念;使用蛋白粒度对氨基酸序列进行分析,进一步给出了蛋白粒度的阶、蛋白粒度的界、蛋白粒度的极限、蛋白粒度增量等概念。主要结论有:1)蛋白粒度等概念和知识能够对蛋白质序列的组成特性进行描述和刻画,蛋白质序列的组成具有粒度偏好;2)蛋白粒度在蛋白质序列上的分布是不均匀的,但没有一个特殊的粒度在蛋白质序列中占绝对优势地位;3)随着蛋白粒度阶的增加,蛋白质在构成序列时更倾向于选择不同的粒度,而不是复用粒度;4)在同阶粒度水平上,蛋白粒度的种类有上界,是个固定值,文中给出了上界值的具体算法;5)每条蛋白质序列都有各自的蛋白粒度种类的极限。
对于蛋白粒度在蛋白质预测中的应用,通过实验证明,蛋白粒度的方法在数据降维上效果明显,在蛋白质序列同源性的高精度区分,以及由于样品不足所形成的小样品的高精度区分上,具有独特优势。
理论分析结论和预测实际结果都表明,蛋白粒度及有关概念的提出是合理的,它从物质凝聚成粒这个角度出发,对氨基酸形成蛋白质序列的过程进行了揭示和刻画,具有一定的生物学理论与实践价值。
1. Chen C,Chen LX,Zou XY,Cai PX.Predicting protein structural class based on multi-features fusion.J Theor Biol,2008,253(2):388~392
2.Wang YC,Wang XB,Yang ZX,Deng NY.Prediction of enzyme subfamily class via pseudo amino acid composition by incorporating the conjoint triad feature. Protein Pept Lett,2010,17(11):1441~1449
3.Xi LL,Li SY,Liu HX,Li JH,Lei BL,Yao XJ.Global and local prediction of protein folding rates based on sequence autocorrelation information.J Theor Biol,2010,264(4):1159~1168
4. Wan SB,Mak MW,Kung SY.mGOASVM:Multi-label protein subcellular localization based on gene ontology and support vector machines.BMC Bioinformatics,2012,13:290.DOI:10.1186/1471-2105-13-290
5. Du PF,Cao SJ,Li YD.SubChlo:Predicting protein subchloroplast locations with pseudo-amino acid composition and the evidence-theoretic K-nearest neighbor(ET-KNN)algorithm.J Theor Biol,2009,261(2):330~335
6. Zhang ZH,Wang ZH,Zhang ZR,Wang YX.A novel method for apoptosis protein subcellular localization prediction combining encoding based on grouped weight and support vector machine.FEBS Letters,2006,580(26):6169~6174
7.林翠琴.组合学与图论.北京:清华大学出版社,2009 Lin CQ.Combinatorics and graph theory.Beijing:Tsinghua University Press,2009
8.Chou KC,Magglora GM.Domain structural class prediction.Protein Eng,1998,11(7):523~538
9.Vapnik V.Statistical learning theory.New York:Wiley-Interscience,1998
10.Brown MPS.Knowledge-based analysis of microarray gene expression data by using support vector machines.Proc Natl Acad Sci USA,2000,97(1):262~267
11.Ding CHQ,Dubchak I.Multi-class protein fold recognition using support vector machines and neural networks.Bioinformatics,2001,17(4):349~358
12.Chen LY,Li QZ.Prediction of the subcellular locatin of apoptosis proteins.J Theor Biol,2007,245(4):775~783
13.Kandaswamy KK,Pugalenthi G,Moller S,Hartmann E,Kalies KU, Suganthan PN, Martinetz T. Prediction of apoptosis protein locations with genetic algorithms and support vector machines through a new mode of pseudo amino acid composition.Protein Pept Lett,2010,17(12):1473~1479
14.Nanni L,Brahnam S,Lumini A.Wavelet images and Chou's pseudo amino acid composition for protein classification.Amino Acids,2012,43(2):657~665
15.Chang CC,Lin CJ.LIBSVM:A library for support vector machines.ACM TIST,2011,2:1~27
16. Chou KC,Zhang CT. Review:Prediction of protein structural classes.Crit Rev Biochem Mol Biol,1995,30(4):275~349
17.Mei SY.Multi-kernel transfer learning based on Chou's PseAAC formulation for protein submitochondria localization.J Theor Biol,2012,293:121~130.DOI:10.1016/j.jtbi.2011.10.015
18.Zhou GP,Doctor K.Subcellular loeation prediction of apoptosis proteins.Proteins:Struct Funct Genet,2003,50(l):44~48
19.Huang WL,Tung CW,Huang HL,Hwang SF,Ho SY.Accurate prediction of enzyme subfamily class using an adaptive fuzzy k-nearest neighbor method. Biosystem,2007,90(2):405~413
20.Shen HB,Chou KC.Predicting protein subnuclear location with optimized evidence-theoretic K-nearest classifier and pseudo amino acid composition.Biochem Biophys Res Commun,2005,337(3):752~756
猜你喜欢
九江职业技术学院学报(2022年1期)2022-12-02
保定学院学报(2022年2期)2022-04-07
粉末冶金技术(2021年3期)2021-07-28
中国洗涤用品工业(2019年4期)2019-05-11
许昌学院学报(2018年4期)2018-05-02
中成药(2018年1期)2018-02-02
中华建设(2017年1期)2017-06-07
中成药(2017年3期)2017-05-17
系统工程与电子技术(2016年12期)2016-12-24
浙江大学学报(工学版)(2016年11期)2016-06-05
